
Document
Ben-Gurion Universityy of the Negev
Beng
Department of Chemistry
MOLECULAR MAGNETISM
AND MATERIALSMATERIALSTHEORY AND APPLICATIONS
Professor Boris Tsukerblat
tsuker@bgu.ac.il
@ g
Beer--Sheva
Beer
2006
SYLLABUS
I. Scope of molecular magnetism. Diversity of the field. Main
kinds of magnetic systems and the main types of the
magnetic
ti ordering.
d i
II. Spin, fundamental equations in molecular magnetism.
Magnetic susceptibility , magnetic moments
moments. Curie-Weiss low
low,
magnetization. Electron paramagnetic resonance.
III. Magnetic properties of a free ion, molecules containing a
unique
i
magnetic
ti center
t without
ith t first-order
fi t d orbital
bit l magnetism
ti
and EPR of transition metal ions and rare-earths, spin-orbital
interaction.
IV. Effects of crystal field. Group-theoretical introduction. Ground
terms of the transition metal ions in the crystal fields.
Anisotropy
py of the g
g-factor. Zero-field splitting:
p
g q
qualitative and
quantitative approaches. Covalence and orbital reduction.
EPR of the metal ions in complexes.
V. Exchange interaction in clusters. Exchange effect, the
nature of the potential exchange. Magnetic properties of
binuclear compounds, dimers of Cu(II) , EPR, magnetic
anisotropy.
VI Heisenberg-Dirac-Van
VI.
Heisenberg Dirac Van Vleck model of the exchange
interaction. Concept of spin-Hamiltonian. Many-electron
problem of the exchange. Spin-coupling scheme for the
polynuclear compounds
compounds, Kambe’s
Kambe s approach.
approach Trimeric
and tetrameric clusters: basic chromium and iron
acetates. EPR spectra of polynuclear compounds.
VII Single
VII.
Si l molecule
l
l magnets, physical
h i l principlesi i l
quantum
tunneling, relaxation. Mn12-ac molecule. Applications in
molecule-based devices.
VIII. Mixed-valence compounds. The phenomenon of
mixed valence. Spin-dependent delocalization-double
exchange- classical and quantum-mechanical
exchange
quantum mechanical
description (Anderson’s theory). Robin and Day
classification of mixed-valence compounds. Intervalence
light absorption (light induced electron transfer)
transfer).
Magnetic properties.
SOURCES
SOURCES:
1) CD
D
(Power Point file)
2)THE MAIN BOOKS AND 3) REFERENCES
THROUGHOUT THE FILE
•
•
•
•
•
Oliver Kahn
Kahn, Molecular Magnetism
Magnetism, VCH,NY(1993).
VCH NY(1993)
Alessandro Bencini, Dante Gatteschi, Electron Paramagnetic
Resonance of Exchange Coupled Systems, Springer-Verlag,
Berlin (1990)
(1990).
F.A.Cotton, Chemical Application of Group Theory,
2nd Edition, Interscience, New York (1971).
B.S.Tsukerblat, Group Theory in Chemistry and Spectroscopy.
A Simple Guide to Advanced Usage, Academic Press, London
(1994).
J.J.Borras-Almenar, J.M.Clemente-Juan, E.Coronado, A.V.Palii,
B.S.Tsukerblat, Magnetic Properties of Mixed-Valence
Clusters:Theoretical Approaches and Applications, in:
“M
“Magnetism:
ti
Molecules
M l
l tto M
Materials”,
t i l ”
( J.Miller, M.Drillon, Eds.), Wiley-VCH (2001) p.p.155-210.
ABOUT THE BOOKS
BOOKS--MOLECULAR MAGNETISM
Chapter 5
ABOUT THE BOOKS
BOOKS--GROUP THEORY
Exceptionally clear presentation !
YOU ARE EXPECTED TO KNOW:
•
•
•
•
Main concept of quantum mechanics: Schrödinger
equation, wave-functions,
f
hydrogen atom, many-electron
atoms, some knowledge of the perturbation theory.
Orbital and spin
p angular
g
momenta. Pauli p
principle.
p
Group theory for chemists (standard course for chemists):
how to determine the point symmetry group that the
molecule belongs to
to, concept of the reducible and
irreducible representations, classification of the molecular
energy levels, selection rules. Classification of molecular
vibrations.
vibrations
Background of the crystal field theory for transition metal
ions: general idea of the crystal field splitting and some
results for the transition and rare
rare-earth
earth ions
ions.
Molecular orbital approach – the main concepts.
YOU ARE EXPECTED TO LEARN
• Magnetic substances.
substances. The main kinds of magnetic
behavior. Basic concepts:
p magnetic
g
moments,, magnetic
g
susceptibility. Spin, free ions, spin-orbit coupling,
g-factors. Electron paramagnetic resonance.
• Crystal
C t l fifield
ld th
theory role
theory,
l off the
th ligands,
li
d magnetic
ti
properties of complex compounds, zero-field splitting,
g
resonance,, anisotropy
py of g
g-factors.
magnetic
• Exchange interaction in clusters. Properties of
polunuclear compounds. Magnetic anisotropy,
nanoscience
i
-single
i l molecular
l
l magnets.
t
• Concept of mixedmixed-valency and electron transfer double
exchange ferromagnetic effect of the double exchange,
exchange,
exchange
role of the electron-vibrational interactions-localization vs.
delocalization. Spin-dependent delocalzation in ironsulphur
l h proteins.
t i
LIST OF THE MAIN NOTATIONS
(to be used as necessary)
• H - magnetic field (vector),
H - magnetic field - absolute value (font --" arial
arial" ).
)
• Vectors and matrices – bold.
• Hˆ Hamiltonia
H ilt i n (font
(f t - " times
ti
new Roman,
R
It li " ).
Italic"
)
• Operators are marked by " cap" : Hˆ , etc .
• S - spin (quantum number), S - classical spin vector,
Sˆ spin
p operator
p
((vector operator
p
- bold and " cap"
p ),
Sˆ x , Sˆ y , Sˆ z components of the vector operator Sˆ .
• L - quantum number of the orbital angular momentum,
momentum
ˆ operator (vector operator).
L - classical vector, L
ˆ.
Lˆx , Lˆ y , Lˆz components of the vector operator L
• J - quantum number of the total angular momentum,
J - classical vector,
ector Jˆ operator (vector
( ector operator),
operator)
Jˆ x , Jˆ y , Jˆ z components of the vector operator Jˆ .
• M S , M L , M J ,- magnetic quantum numbers of spin, orbital
angular momentum and total angular momentum.
• g e g factor of a free electron,
g J g factor of a LSJ state.
• - magnetic susceptibi lity.
• μ magnetic
g
moment ((classical vector),
),
ˆ magnetic moment (vector operator),
• μ
magnetic moment (absolute value)
• Hˆ ZFS zero field splitting Hamiltonia n.
• D zero - field splitting parameter.
parameter
• J exchange parameter
Chapter
p I
Scope of molecular magnetism.
Diversity of the field. The main kinds of
magnetic systems and the main types
of the magnetic ordering
SCOPE OF MOLECULAR MAGNETISM
♠ Magnetic properties of isolated atoms ,ions
ions and
molecules ( in particular, metal complexes) containing
one magnetic
g
center.
Example: complex ion –coordination compound
Metal--(Ligand)
Metal
( g
)6
NH3
NH3
NH3
Cr
NH3
NH3
NH3
[Cr (NH3)6]3+ or
[Cr(NH3)6]Cl3
Central metal ion
ion- Cr3+
surrounded by six ammonia
molecules, Cr3+ contains three
unpaired d-electrons
Octahedral symmetry, Oh point group
♠ Magnetic properties of the molecules containing more
than one magnetic centers – polynuclear compounds,
magnetic clusters or exchange clusters.
Example: binuclear Co cluster, bi-octahedral edgeshared geometry- oxygen bridged system
NH3
NH3
NH3
OH Co
NH3
Co
OH
NH3
NH3
NH3
[(H3N)4Co(OH)2Co(NH3)4]4+
NH3
Co2+(d7-shell)C
h ll)
bearer of
magnetism
Point symmetry D2h
POLYOXOANION [Ni3 Na(H2O)2(AsW9O34]11WO6
NiO6
3Ni2+magnetic
g
fragment
Na
AsO4
Polyhedral representation
Ball and stick representation
Inorg. Chem. ,2003, 42, 5143-52
POLYOXOANION [Ni6 As3W24O94(H2O)17]-
Two 3Ni2+T
magnetic
f
fragments
t
WO6
NiO6
AsO4
P l h d l representation
Polyhedral
t ti
B ll and
Ball
d stick
ti k representation
t ti
Inorg. Chem. ,2003, 42, 5143-52
♠ Assemblies of molecules with the magnetic interactions
b t
between
th
the molecular
l
l entities,
titi
one-dimensional
di
i
l systems
t
Structure of donor-acceptor
compound (TTF)+[CuS4C4(CF3)4]with TTF+= tetrathiafulvalinium.
Phys.Rev.Lett.,35(1975) 744
Structure of the ferrimagnetic chain
MnCu(pba)(H2O)3·2(H2O) with
pba=
b 1,3-propilene-bis(oxamato).
13
il
bi (
)
Inorg.Chem.,26(1987)138.
DIVERSITY OF THE FIELD,SELECTED
APPLICATIONS
L
N
Material
sciences
Biology
Molecular
magnetism
Molecular
M
l
l
electronics
NanoN
Nanoscience
SINGLE MOLECULAR MAGNETMAGNETMAGNET IN ONE MOLECULE
[Mn12O12 (CH3COO)16 (H2O)4] -molecule - Mn12-ac (Mn12-acetate)
Molecular
electronics
NanoNanoscience
i
Mn4+
Mn 3+
MANGANESE--12 CLUSTER
MANGANESE
eight Mn3+ ions (Si =2) and four Mn4+(Si =3/2)
PHYSICAL BACKGROUND – BRIEFLY
Pictures: Michel Verdaguer
z
Thermal activation
E
y
x
En
nergy
0
2
DS
z
DS2
- Sz
Magnetization vectors
-4 -2 0 +2 +4
Direction of magnetization
Sz
+Sz
Barrier for anisotropy
If tthe
e Mn12-ac
ac molecule
o ecu e is
s magnetized
ag et ed by a
an app
applied
ed
field, the molecule retains magnetization for a long time ,
approximately 108 seconds = 3 years at 1.5K
A li ti
Applications:
quantum computing , memory storage elements in one molecule
MULTIFUNCTIONAL MATERIALS
Molecular
electronics
Material
sciences
Nature 408, 421 - 422 (2000)
Molecular electronics: A dual
dual-action
action material
FERNA NDO PALACIO* AND JOE L S. MILLE R
Fernando Palacio is at the Instituto de Ciencia de Materiales de Aragón, CSIC, Universidad de
Zaragoza, 50009 Zaragoza, Spain.
e-mail: palacio@posta.unizar.es
Joel S. Miller is in the Department of Chemistry, University of Utah, Salt Lake City, Utah 841120850, USA.
e-mail: jsmiller@chemistry.utah.edu
In the drive for smaller electronic components, chemists are thinking on a
molecular
l
l scale.
l By
B combining
bi i two simple
i
l molecules,
l
l
a hybrid
h b id has
h been
b
produced
d
d
that is both magnetic and an electrical conductor.
DISCOVERY OF MULTIFUNCTIONAL
MOLECULE--BASED MATERIALS
MOLECULE
Nature 408, 447 - 449 (2000)
Coexistence of ferromagnetism and metallic
conductivity in a molecule-based layered compound
EUGENIO CORONADO*, JOSÉ
É R. GALÁN-MASCARÓS*†,
Á
Ó
CARLOS J. GÓME
Ó
Z-GA RCÍA*
Í &
VLADIMIR LAUKHIN*†
* Instituto de Ciencia Molecular, Universidad de Valenc ia, Dr. Moliner 50, 46100 Bur jjasot, Spain
p
† Present addresses: Department of Chemistry, Texas A&M University, College Station, Texas, USA (J.R.G.- M.); ICMBCSIC, Campus de la UAB, 08193 Bellaterra, Spain (V.L.)
Crystal engineering
engineering—the
the planning and construction of crystalline supramolecular
architectures from modular building blocks—permits the rational design of functional
molecular materials that exhibit technologically useful behavior such as conductivity
and superconductivity,
superconductivity ferromagnetism and nonlinear optical properties.
properties Because the
presence of two cooperative properties in the same crystal lattice might result in new
physical phenomena and novel applications, a particularly attractive goal is the design
of molecular materials with two properties that are difficult or impossible to combine
in a conventional inorganic solid with a continuous lattice.
A DUAL ACTION MATERIAL
Molecular
components:
( ) The
(a)
organic
molecule
BEDT TTF
BEDT-TTF
bis(ethylenedithio)
tetrathiafulvalene
(b) A ferromagnetic bimetallic complex of
manganese(ii)tris(oxalato)chromium(iii).
Carbon atoms are in p
pink, sulphur
p
in blue.
By alternating layers of the molecules in a and b, E. Coronado et al. (Nature)
have created a hybrid material that supports both magnetism and conduction.
M- magnetic layers, E-conducting layers
Structures of the hybrid material and the two sublattices.
sublattices
a, View of the [MIIMIII(C2O4)3]- bimetallic layers. Filled and open circles in the
vertices of the hexagons represent the two types of metals.
b Structure
b,
St t
off the
th organic
i layer,
l
showing
h i the
th packing
ki off th
the BEDT
BEDT-TTF
TTF molecules.
l l
c, Representation of the hybrid structure along the c axis,
showing the alternating organic/inorganic layers.
BIOLOGICAL SYSTEMSSYSTEMS-TWO EXAMPLES
Tri--iron cluster
Tri
Di--iron cluster
Di
S-cys
S
Fe
Schematic structure of the
protein with [Fe3S4] core.
core
S-cys stands for the sulfur atom
of a cystein group.
Th
Three
magnetically
ti ll coupled
l dF
Fe iions.
Schematic structure of the
two iron (Fe2+, Fe3+ ) ferredoxin
two-iron
ferredoxin.
S-cys stands for the sulfur atom
of a cystein group.
T
Two
magnetically
ti ll coupled
l dF
Fe iions.
MAIN KINDS OF MAGNETIC SUBSTANCES
paramagnet
ferromagnet
antiferromagnet
Disordered
directions of
the magnetic
moments,,
macroscopic
magnetization
is zero
Long-range
collinear
alignment of all
moments in the
substance,
spontaneous
magnetization
Long-range
interaction
interaction,
moments are
aligned
antiparallel to
each other, no
magnetization
ferrimagnet
Antiparallel
different
magnetic
moments,
spontaneous
macroscopic
magnetization
weak ferromagnet triangular structure
Two sub-lattices
with non-collinear
non collinear
magnetic
moments, weak
spontaneous
magnetization
Triangular
configuration
of the
magnetic
g
moments
HELICOIDAL MAGNETIC STRUCTURES
simple helix
ferromagnetic
helix
complex
helix
static
longitudinal
spin wave
SCHEMATIC PHASE DIAGRAM OF BULK
HOLMIUM
Chapter II
Spin, fundamental equations in
molecular magnetism.
Magnetic susceptibility , magnetic
moments, electron paramagnetic
resonance
MAGNETIC FIELD, MAGNET IN A FIELD
permanent magnet
North
N
S
South
Magnetic
g
field H
F
N
F
Turning moment acting on a magnetic stick
in a homogeneous magnetic field
SPIN OF THE ELECTRON
Electron - elementary bearer of magnetism
Elementary magnetic moment :
|e|
0.9284 10 20 erg gauss 1 0.92740 10 24 J T 1
2mc
Borh magneton, two convention ally accepted notations :
or μ B
h
1.05 10 27 erg s , h Planck constant
2
e 4.8 10 10 cgse charge of the electron,
vacuum
c 3 1010 cm s 1 velocity of the light in vacuum,
m 9.1 10 28 g mass of the electron,
1T 1Tesla 10 4 gauss
SPIN--BEARER OF THE MAGNETIC MOMENT
SPIN
Classical image
image-rotating
rotating spherical charge,
this picture fails in the evaluation of spin magnetic moment.
Adequate
q
description
p
-q
quantum--mechanical
quantum
concept.
MAGNETIC MOMENT
Magnetic moment associated with the spin (mechanical
angular momentum) of the electron can have two projections
on the direction of the external magnetic field H:
Magnetic
field H
S
S
e
2mc
spin “down” S
e
2mc
N
N
S
spin “up”
up
S
e
2mc
ELEMENTS OF QUANTUM MECHANICS OF SPIN
Notation for the spin function
S , M
S spin, M S quantum number of spin projection
Sˆ spin operator (notation " cap" ) - operator of spin angular momentum
Vector operator Sˆ , Sˆ , Sˆ three components !
x
y
z
GENERAL PROPERTIES :
Sˆ 2 S , M S S S 1 S , M S
Sˆ S , M M S , M
z
S
S
S
Spin - functions are the eigen - functions of two operators :
Sˆ 2 and Sˆ
z
with the eigen values : S S 1 of Sˆ 2 and M S of Sˆ z
M S quantum number of spin projection ,
M S S , S 1, , S 2 S 1 values - spin multiplicity
THE CASE OF SPIN S
S=1/2
1/2
Two spin projection s : ms
1
2
and ms 12
Spin wave functions s , ms 12 , 12 and 12 , 12
Sh t notations
Short
t ti
f s 12 :
for
spin " up
up" and spin " down
down"
ˆs operator of spin - 21 , s s 1 12 32
Main properties :
ˆs 2 34 , ˆs 2 43
ˆs z 12 , ˆs z 12
3
4
SPATIAL QUANTIZATION - AN IMPRESSIVE RESULT
OF QUANTUM MECHANICS - PHYSICAL PICTURE
MS
1
2
MS
MS 1
3
2
MS
1
2
MS 0
M S 12
M S 12
S
1
2
M S 1
M S 32
S 1
S
3
2
Classical
C
ass ca mechanics
ec a cs ►all
a d
directions
ect o s for
o tthe
e magnetic
ag et c
moment in the space are allowed.
allowed
Quantum mechanics ► only selected directions for the
magnetic moment in the space are allowed
allowed-spatial quantization. Arbitrary z-axis.
VECTOR (CLASSICAL) MODEL FOR THE ANGULAR
MOMENTA IN QUANTUM MECHANICS
Z
Vector S precesses around
arbitrary direction Z at the
conical surface
surface, so that the mean
values of the projections of S at
the p
plane p
perpendicular
p
to axis of
Z are zero (SX , SY).
“Good” quantum numbers:
S and MS
SZ
S
θ
S S S 1
2
SY
squared
d absolute
b l t value
l
SX
(length) of the vector S.
φ
Y
Spatial quantization►
X
Classical picture
selected directions θ(MS):
MS
Mean values: <SX > = <SY> =00
cos
S S 1
<SZ> = S·cosθ
PRECESSING SPIN –
CLASSICAL PICTURE ILLUSTRATING
THAT:
Z
mean values
<SX > = <SY> =0,
0
but <SZ> = S·cosθ 0
X
Y
Vector S performs precession around arbitrary direction Z at
the conical
surface in an external magnetic field the
surface,
precession occurs around the vector of external magnetic field
IImage from:
f
h //
http://www.weizmann.ac.il/chemphys/Vega_group/home.html
i
il/ h
h /V
/h
h l
Prof. Shimon Vega , Weizmann Institute of Science, Israel
SPATIAL QUANTIZATION:
ILLUSTRATION for S=1/2
Z
MS
1
2
S
projections
S 12 M S 12 and M S 12
S S S 1
54.7
MS
cos
S S 1
125.3
M S 12
cos
S
1
3
M S 12 cos
M S 12
S
1
2
3
2
54.7
1
3
125.3
SPATIAL QUANTIZATION:
ILLUSTRATION for S=1
Z
S 1 M S 1 , M S 0 , M S 1
MS 1
45
S 2
S
cos
MS 0
90
135
M S 1
MS
S S 1
M S 1 cos
1
2
45
M S 0 cos 0 90
M S 1 cos
S 1
1
2
135
ZEEMAN INTERACTION interaction of the electronic spin
p with the magnetic
g
field
Operator of spin magnetic moment :
ˆ S g e ˆs
μ
Vector operator (three components ) :
ˆ x 2 β ˆs x , ˆ y 2 β ˆs y , ˆ z 2 β ˆs z
g e factor Lande, or g - factor for a free electron :
g e 2.0023 2
Energy of the interactio n of the magnetic moment μ
with the external magnetic field H :
E Z μH x H x y H y y H y
Zeeman Hamiltonia n :
Hˆ Z g e ˆsH
ZEEMAN INTERACTIONINTERACTION- ARBITRARY SPIN S>1/2
ˆ S g e Sˆ ,
μ
Sˆ ˆsi total spin of an atom or ion
i
Summation of the vectors ˆsi
over all unpaired electrons
( numbered by the symbol " i " ) in the atomic shell
Interactio n of the magnetic moment with the magnetic field H :
E μ H μ H cos , angle between vectors μ and H.
Hamiltonia n can be obtained
by substituti ng classical values by their operators :
ˆ
E Hˆ , μ μ
Z
Hˆ Z g e Sˆ H g e Sˆ xH x Sˆ yH y Sˆ zH z
ZEEMAN INTERACTION FOR A SPIN STATE - S
H
θ
μ
Notation for the magnetic field :
H (vector-"
t " bold
b ld" ),
) Hx , Hy , Hz projection
j ti s (scalars).
(
l )
Hamiltonia n of Zeeman interaction
(magnetic field along z - axis, H x H y 0, H z 0) :
Hˆ Z g e Sˆ zHz
Important
p
remark:
z-axis is chosen arbitrary, free atom is spherically symmetric
ZEEMAN LEVELS FOR A SPIN STATE - S
S h di
Schrodinge
r equation
ti :
Hˆ SM E SM ,
Z
S
S
g e Hz Sˆ z SM S g e Hz M S SM S
S li i under
Splitting
d the
h action
i off magnetic
i field
fi ld
(Zeeman levels) :
E M S g e H z M S
2S 1
1 eigen - values
Magnetic field removes (2S+1)-fold degeneracy of spin level
For a free atom ( ion) Zeeman splitting is independent of
the direction of the field - isotropic in space
Paul Maurice Adrien
Dirac
1960
English theoretical physicist known for his work in quantum mechanics and
for his theory of the electronic spin . In 1933 he shared the Nobel Prize with
the Austrian physicist Erwin Schrödinger .
ZEEMAN SPLITTING FOR A FREE ELECTRON
E mS g e Hz mS , mS 12
E 12 12 g e Hz
H 0 E 12 E 12
H 0 E 12 E 12
ene
ergy
E(mS)
mS 12
S
N
S 1/2
S=1/2
mS
1
2
splitting
mS
magnetic field Hz
1
2
N
S
Magnetic field removes (2S+1)-fold degeneracy of a spin level:
energy levels become dependent of spin projections mS
Pieter Zeeman
Born May 25, 1865,
Zonnemaire, Netherland.
Died Oct.
Oct 9,
9 1943,
1943 Amsterdam
Nobel Winner, 1903 :
for his discoveryy of the Zeeman effect
Zeeman effect in physics and astronomy,
astronomy the splitting of a spectral line into
two or more components of slightly different frequency when the light source
is placed in a magnetic field
field. It was first observed in 1896 by the Dutch
physicist
h i i t Pieter
Pi t Zeeman
Z
as a broadening
b d i off the
th yellow
ll
D li
D-lines
off sodium
di
i a
in
flame held between strong magnetic poles. Later the broadening was found to
be a distinct splitting of spectral lines into as many as 15 components.
Zeeman's discovery earned him the 1902 Nobel Prize for Physics, which he
shared with a former teacher, Hendrik Antoon Lorentz, another Dutch
pphysicist.
y
Lorentz,, who had earlier developed
p a theory
y concerning
g the effect of
magnetism on light, hypothesized that the oscillations of electrons inside an
atom produce light and that a magnetic field would affect the oscillations and
thereby the frequency of the light emitted. This theory was confirmed by
Zeeman's research and later modified by quantum mechanics, according to
which spectral lines of light are emitted when electrons change from one
discrete energy level to another.
another Each of the levels,
levels characterized by an
angular momentum (quantity related to mass and spin), is split in a magnetic
field into substates of equal energy. These substates of energy are revealed by
th resulting
the
lti patterns
tt
off spectral
t l line
li components.
t
Pieter Zeeman, Albert Einstein,
Paul Erenfest
Pieter Zeeman and Niels Borh
Magnet of
Pieter Zeeman
John H.
H Van Vleck
American physicist and mathematician who shared the Nobel Prize for Physics in
1977 with Philip W. Anderson and Sir Nevill F. Mott. The prize honoured Van Vleck's
contributions to the understanding of the behaviour of electrons in magnetic,
noncrystalline solid materials.
Van Vleck developed during the early 1930s the first fully articulated quantum
mechanical
h i l theory
th
off magnetism.
ti
L t he
Later
h was a chief
hi f architect
hit t off the
th ligand
li
d field
fi ld
theory of molecular bonding. He contributed also to studies of the spectra of free
molecules, of paramagnetic relaxation, and other topics. His publications include
Q ant m Principles and Line Spectra (1926) and the Theory
Quantum
Theor of Electric and
Magnetic Susceptibilities (1932).
ZEEMAN SPLITTING,
ILLUSTRATION FOR SPIN S=1
E M S g e Hz M S , M S 1, 0 , 1
Enerrgy
M S 1 E 1 g e Hz
S 1
M S 0 E 0 0
M S 1 E 1 g e Hz
M
Magnetic
ti fi
field
ld
ELECTRON PARAMAGNETIC RESONANCERESONANCECLASSICAL PICTURE
Zeeman splitting- constant magnetic field along Z-axis.
Alternating magnetic field in the XY plane:
H X t H X cos t , 2 cyclic frequency of the field
frequency of the alternatin g field,
1 , period (time of one cycle)
H0
Z H0-constant field
Hr-rotating
g field
force
Y
X
Rotating field produces a turning momentmoment
to align spin in the plane XY, i.e. parallel to Hr !
precessing
spin
force
CONDITION FOR THE RESONANCE
Constant field
Resonance condition:
frequency of rotating field=
frequency of spin precession
H0
0
turning
moment
Hr
Rotating field
Electron paramagnetic
resonance (EPR), or electron
spin
i resonance (ESR) .
Eugenii Zavoisky, Kazan, 1944
Frequency of precession
in the constant field H0 :
g H0
g H0
0
or 0
h
Under the resonance
condition the turning
g
moment acts in-phase with
spin precession and spin
rapidly changes orientation.
CLASSICAL PICTURE OF THE
ELECTRON PARAMAGNETIC
RESONANCE
H X cos t
Spin “up”
up
Spin “down”
down
ROTATING “PERPENDICULAR“ MAGNETIC FIELD
OF THE RESONACE FREQUENCY
REVERSES SPIN
QUANTUM DESCRIPTION OF EPR
E
Zeeman interaction
S=1/2
M S 12
EPR
transition
M S 12
H0
with the alternating field
ˆ g Sˆ H cos t .
H
alt
X
X
This interaction induces transitions
M S M S
between different Zeeman levels E M S :
E M S g H0 M S
Important rule -" selection rule" for quantum transitions
b t
between
Z
Zeeman
l
levels
l :
only the transitions between " neighborin g" Zeeman levels are allowed
M S M S 1 and
d M S M S 1 or M S M S 1
Resonance condition (energy conservati on low) :
increase of spin energy energy of quantum of alternating field
E M S E M S 1
QUANTUM RESONANCE CONDITION
(arbitrary spin )
E M S E M S 1
E M S g H M S for
f an arbitrary
bit
spin
i value
l S
g H M S g H M S 1 g H0
Th resonance condition
The
di i within
i hi quantum - mechanical
h i l approach
h:
g H0
g H0 quantum energy for an allowed transition,
But
g H0
0 cyclic frequency of classical
spin
p p
precession in magnetic
g
field H0 .
The main conclusion :
quantum frequency for an allowed transition
cyclic frequency of spin precession in magnetic field
DETECTION OF RESONANT ABSORPTION
Some estimations of the physical values:
for a free electron (g=2) at frequency of 30GHz (Gigahertz)
(1GHz=109Hz) the resonant field H0=10,700Gauss.
30GHz-area of microwave frequencies of radiation,
energy ≈ 1cm
1 -11 (1ev
(1 = 8,066cm
8 066 -11).
)
Case I: the separation of the Zeeman levels is fixed by holding
the magnetic field constant; the microwave frequency ω is then
varied until a resonance absorption is found.
ω
g H0
0
Resonance: ω= ω0
DETECTION OF RESONANT ABSORPTION
Case II: the microwave frequency is fixed; the magnetic field is
then varied. The characteristic aspect of EPR spectroscopy is
the variation of the energy
gy level separation
p
by
y variation of the
magnetic field until the resonance is reached (at H=Hres ).
non-resonance
frequences
resonance frequency
12 g H
g Hres
ω
12 g H
EPR line
Hres
H
Resonance equation:
Hres resonance field
Characteri zation of g - factor :
g eff
Hres
Preliminary remark: g=2 only for a free electron
EPR, S>1/2 - ISOTROPIC SYSTEM
E M S g H0 M S
3
S
2
Forbidden
t
transitions
iti
3
2
g H0
1
2
g H0
1
2
g H0
3
2
g H0
Hres H0
EPR line
In the case of
S>1/2 all allowed
transitions have the
same resonance
fields and for this
reason give the
only
l EPR line.
line
li
Thi
This
is valid in the case
of isotropic Zeeman
interaction (free
atoms or the case
of a cubic crystal
field).
RESONANCE FIELDS AND g
g--FACTORS
Allowed transitions M S M S 1, resonance condition :
g Hres M S 1 g Hres M S
g Hres
g
Hres
Forbidden transitions M S M S 2 and M S M S 3,
two resonance conditions :
1) g Hres M S 2 g Hres M S
2 g Hres
g
2 Hres
and
2) g Hres M S 3 g Hres M S
3 g Hres
g
3 Hres
These two lines in " low fields" can not be observed
MAGNETIZATION OF A SUBSTANCE
A single spin- “up” or “down” →
mS 12
mS 12
Ensemble of N non-interacting spins in a
magnetic
ti fifield
ld ((spins
i ““up”” and
d “d
“down”):
”)
N↑- number of spins “up”, N↓- number of spins “down”
N↑+N↓=N
Magnetic moment of N spins :
N N N N
Main question: numbers N↑ and N↓ - ?
BOLTZMAN DISTRIBUTION
molecules in the medium (ensemble):
(each molecule having, let say, three levels):
3
2
1
Due to interaction with the medium (thermostat or bath) electrons
(bolls-) jump “up” (absorption of heat) and “down” (emission of
heat) traveling among the levels 1,
1 2 and 3.
3 These jumps are very
fast, so one can say about the distribution of the electrons over the
levels in the thermodynamic equilibrium of the ensemble.
N1-mean number of the molecules with the energy E1
N2 -mean number of the molecules with the energy E2
N3 -mean number
b off th
the molecules
l
l with
ith th
the energy E3
N= N1+ N2+ N3-total number of the molecules.
p1= N1/N- p
probability
y to find a molecule with the
populated level 1 (i.e. with the energy E1), etc…
The main question: what are these probabilities?
BOLTZMAN DISTRIBUTIONDISTRIBUTIONGENERAL EXPRESSION
Probability to find a molecule with the populated level “ i ”
(i e a molecule with the energy Ei):
(i.e.
1 Ei / kT
pi e
Z
k B lt
k-Boltzman
constant
t t, T-absolute
T b l t temperature
t
t ,
Z-partition function (important characteristics !!!)
Z e Ei
kT
summation over
all levels
i
Probability pi depends on
the energy Ei and on the temperature T
BOLTZMAN DISTRUBUTIONDISTRUBUTIONILLUSTRATION FOR TWO LEVELS
2
E excited
E
0 ground
1
p1
1
E
1 e kT
, p2
e
E
kT
E
1 e kT
onlyy
E
kT
Z 1 e
E1 0 , E2 E
energy
gy is counted
from the ground level
T 0, p1 1, p2 0
level "1" is p
populated
p
1
T , p1 p2
2
(levels "1" and "2" are equally populated)
Ene
ergy E
POPULATION OF THE ENERGY LEVELS IN
THE THERMODYNAMIC EQUILIBTIUM
E5
1 Ei
pi T e
Z
E4
E3
E2
E1
0
population
kT
p
1 decreases with the increase of the
• Population exponentially
energy; level “i” is populated significantly if kT ≥ Ei ;
• The ground level is always (at any T )
the most populated level .
PARTITION FUNCTION FOR A SPIN S IN A
MAGNETIC FIELD
Z e
i
Ei kT
E M S g e HM S , M S S , ..., S
H - magnetic field, arbitrary orientation,
magnetical ly isotropic system
S
Z exp g e HM S kT
S
Final result ( after the summation is made) :
Sinh 2 S 1x 2
Z
,
Sinh x 2
Hyperbolic sine :
ge H
x
kT
Sinh 12 e e
Hyperbolic cosine : Cosh 12 e e
MAGNETIZATION –QUANTUM
QUANTUM--MECHANICAL
EXPRESSION
In classical mechanics,
when a sample is perturbed by an external magnetic
field, its magnetizat ion is related to its energy variation through
E
M
H
Using
g the language
g g of q
quantum mechanics
we consider a molecule with an energy spectrum
E i 1, 2 ,,...
in the presence of a magnetic field H.
For each energy level we can define
i
a microscopi c magnetizat ion i as
Ei
i
H
MAGNETIZATION –QUANTUM
QUANTUM--MECHANICAL
EXPRESSION, MEAN VALUE
The macroscopi c molar magnetizat ion M is
then obtained by summing up
the microscopi c magnetizat ions
averaged according to the Boltzman
distribution low ( N Avogadro'
g
s number)) :
N exp E kT
M
exp E kT
i
i
i
i
i
and then
N E H exp E kT
M
exp E kT
i
i
i
i
i
This is a fundamenta l expression in molecular magnetism
MAGNETIC SUSCEPTIBILITY
Molar magnetic susceptibility (isotropic system) :
M
M H, or
H
Expressions through the partition function :
N i Ei H exp Ei kT
ln
l Z
1 kT
H
i exp Ei kT
Ei functions
f
ti
off the
th magnetic
ti field,
fi ld Ei Ei H
This leads to the following expressions for magnetizat ion and
magnetic
i susceptibi
ibility
li through
h
h derivative
d i i s off the
h partition
i i function
f
i :
ln Z
M N kT
H
2 ln Z M
N kT
2
H
H
CALCULATION OF THE MOLAR MAGNETIZATION
Sinh 2 S 1x 2
Z
,
Sinh x 2
ln Z g e
H
2kT
ge H
x
kT
2S 1Coth 2S 1x 2 Coth x 2
This can be rewritten as :
ln Z g e
H
2kT
2S 1Coth 2S 1 y
2 S Coth y 2 S
with
ge S H
y
kT
Cosh e e
Hyperbolic cotangent : Coth
Sinh e e
MOLAR MAGNETIZATION
Th molar
The
l magnetizat
ti t ion
i is
i then
th :
ge SH
M Ng
N e S BS y , y
kT
BS y is the Brillouin function defined as :
2S 1
2S 1 1
1
BS y
Coth
y
Coth
y
2S
2S
2S
2S
Cosh e e
Coth
Sinh e e
Two extreme cases for the Brillouin function :
1) y 1, low temperatur e, kT Zeeman splitting
2) y 1, high temperatur e, kT Zeeman splitting
BRILLOUIN FUNCTIONFUNCTIONFIELD AND TEMPERATURE DEPENDENCE
BS
1
S
7/2
5/2
0
3/2
1/2
ge H
kT
• Low temperature or/and high magnetic field: BS → 1
• High
Hi h temperature or/and
/ d weak
k magnetic
i fifield:
ld BS → 0
MAGNETIZATION – LOW TEMPERATURE LIMIT
When T→0 or field is strong , y=gβSH/kT becomes large ,
BS(y) tends to unity.
L
Low
temperature (hi
(high
h fifield)
ld) lilimit
i off molar
l magnetization:
i i
Msat M T 0 Ng e S N 2 S
g e 2
This is the saturation value –only ground Zeeman
l
level
l MS=-SS is
i populated:
l t d
Maximum magnetization
magnetization-all
all spins along magnetic field
1.38 10 23 J K
g H
H
T
4
.
1
0
7
,
1
T
(T
(Tesla)
l
)
10
gauss
27 J
kT
T 2 9.27 10
K
T
MOLECULAR MAGNETSMAGNETSSPIN ALIGNMENT IN EXTERNAL
MAGNETIC
MAGNE IC FIELD
H
Paramagnetic-disordered
g
Ordered (p
(parallel to field))
SATURATION OF MAGNETIZATION, S=1/2
MS
1/2
1/T
-1/2
E 12 12 g e Hz
E 12 12 g e Hz
H
PHYSICAL SENCE OF SATURATION (S=1/2)
E 12 12 g e Hz
E 12 12 g e Hz
Boltzman factors for two Zeeman sublevels:
p 1
2
1
ge H
1 e kT
, p1
2
ge H
e kT
ge H
1 e kT
Decrease of T ( at fixed field –fixed energy gap) increases
population of the ground level.
Increase of magnetic field ( at a certain T) increases the
Zeeman gap and thus increases population of the ground
l
level
l and
dd
decreases population
l ti off th
the excited
it d llevel.l
MAGNETIZATION – HIGH TEMPERATURE LIMIT
We can check that for small
ll y=gβH/kT , BS(y) may be
replaced by the first term of the expansion in terms of y :
BS y y S 1 3S terms in y ...,
3
g SH
y
kT
g SH
smallll y
1 high
hi h temperatur
t
t e and/or
d/ low
l
fi ld
field
kT
(under standard experimental conditions this means T 1 5K )
g SH S 1
BS y
kT
3S
Molar magnetization in this limit :
ge S H S 1
M Ng SBS y Ng S
3S
kT
Ng 2 2
M
S S 1 H
3kT
k
In all expressions : g e g (more general)
FIELD DEPENDENCE OF MAGNETIZATIONMAGNETIZATIONCLASSICAL PICTURE
Ng 2 2
M
S S 1 H
3kT
Weak field
disordered
Applied magnetic field
partially
p
y ordered
Strong
g field
fullyy ordered
MAGNETIC SUSCEPTIBILITYSUSCEPTIBILITY-CURIE LAW
Ng 2 2
N
M
S S 1 H
3kT
Magnetic susceptibility :
M
Ng 2 2
S S 1
H
3kT
Magnetic susceptibility varies as a function of temperatur e C
T
:
C
Ng 2 2
S S 1
, C
T
3k
This is the Curie low which was proposed in 1910 from experiment al
data before the introduction of quantum mechanics.
mechanics
Experiment al verification :
to measure T as a function T , this should be a horisontal straight line :
T C
EFFECTIVE MAGNETIC MOMENTS
Ng 2 2
N
S S 1
3kT
The value g 2 2 S S 1 is the squared value of the
magnetic moment for a particle with spin S :
2 g 2 2 S S 1
Experiment al magnetic susceptibility data can be presented
in the form of the temperatur e dependence of the so - called
effective magnetic
g
moment :
effff 3kT
1
2
N 2
In cgsemu units N 2 3k 0.12505, very close to 1 .
8
eff 8T
1
2
EXPLANATIONEXPLANATIONQUANTUM MECHANICAL BACKGROUND
Operator
p
of the magnetic
g
moment :
ˆ g Sˆ μ
ˆ 2 g 2 2 Sˆ 2
μ
S
S
Accordingly to the rule off quantum mechanics
the mean value of the
physical quantity A in a quantum state
with
ith the
th wave - function
f
ti n r
ˆ operator of A :
should be calculated as A
ˆ r d n A
ˆ n Dirac' s notation
A r A
n
n
MAGNETIC MOMENT
The mean value of squared magnetic moment of a particle
with spin S should be calculated as :
2 g 2 2 SM Sˆ 2 SM
S
S
S
SM S are the eigen - functions of Sˆ 2
Sˆ 2 SM S S S 1 SM S
2S g 2 2 SM S S S 1 SM S
g 2 2 S S 1 SM S SM S
SM S SM S 1 normalization condition
Final result :
2S g 2 2 S S 1 or S g S S 1
MAGNETIZATIONMAGNETIZATIONFIELD AND TEMPERATURE DEPENDENCE
Magnetization M in N units versus H kT plots for
molecules possessing ground state with spin S , g e 2
Pierre Curie
Curie,
1903 Nobel Laureate
in Physics
MAGNETIC MOMENTS OF SOME METAL IONS
Maagnetic momeent/mol, β
Gd3+, S=7/2- Gd-sulphate
Fe3+, S=5/2- iron-ammonium
alum
Cr3+, S=3/2- chromium-potash
alum
l
Brillouin
Experimental data:
Henry W., Phys.Rev.,88 (1952) 559
H/T, Tesla/K
• Strong field and/or low temperature ► Msat=2βS
• Weak
W k field
fi ld and/or
d/ high
hi h temperature ► M=0
Chapter III
Magnetic properties of a free ion,
molecules containing a unique magnetic center
without first-order
first order orbital magnetism and EPR of
transition metal ions and rare-earths, spin-orbital
interaction.
interaction
QUANTUM--MECHANICAL DESCRIPTION
QUANTUM
OF A FREE ATOM
Quantum numbers, spinspin-orbital
coupling, gg-factors
Henrik David Bohr Niels
Erwin Schrodinger
Born: 12 Aug 1887 in Erdberg, Vienna, Austria
Died: 4 Jan 1961 in Vienna,, Austria
Nobel Prize, 1933 –fundamentals of
QUANTUM MECHANICS
Wolfgang Ernst Pauli , Born: 25 April 1900 in Vienna, Austria
Di d 15 D
Died:
Dec 1958 in
i Zurich,
Z i h Switzerland
S it l d
In 1945 he was awarded the Nobel Prize for decisive contribution through his discovery in
1925 of a new low of Nature, Pauli exclusion principle. He had been nominated for the
Prize by Albert Einstein
QUANTUM NUMBERS
FOR ONE ELECTRON IN A SPHERICAL POTENTIAL
(HYDROGEN ATOM,
ATOM ONE ELECTRON IONS)
n the main quantum number n 1,2,3...
l quantum number of the orbital
angular momentum l 0, 1,...,n 1
ml magnetic quantum number
ml l , l 1,...,l 1, l 2l 1 values
l
ms spin projection quantum number
spin
s 1 2, ms 1 2 :
spin " up
up"- and " down
down"-
parity of the quantum state, 1 l,
" even" and " odd" states, p-" odd" , d - even, etc.
SPECTROSCOPIC NOTATIONS:
l 0 , 1, 2 , 3, 4 , 5 ,...
s
p
d
f g
h
“even”
even and “odd”
odd states:
s–even (l=2), p-odd ( l =1), d –even (l=2)
TRANSITION METAL IONS
Ions of transition
metals of the iron
group have unfilled
3d – shells:
n 3, l 2
Closed d-shell
contains 10 electrons:
(2l+1)·2=10
Typical oxidation degrees and d n:
d Ti
1
3
,
d V
2
3
d 3 Cr 3 ,V 2 ,
d
4
d
5
d7
d
9
,
Mn , Cr ,
Fe , d Fe ,
Co , d Ni ,
Cu
3
3
2
2
2
2
6
8
2
ATOMIC TERMS, SPECTROSCOPIC NOTATIONS
DEFINITION: 2S+1L
(or SL)-TERMS
closed
shells
unfilled
shells
Rule of the addition of the
angular (spin and orbital)
momenta
(vector coupling scheme):
L l1 l2 , l1 l2 1,....,|l1 l2 |
S s1 s2 , s1 s2 1,...,| s1 s2 |
L QUANTUM NUMBER OF THE
ORBITAL ANGULAR MOMENTUM
OF THE ELECTRONIC SHELL
S FULL SPIN OF THE
ELECTRONIC SHELL
3
P L 1, S 1 ,
F L 3, S 3 2
4
GROUND TERMS OF TRANSITION METAL IONS
IONS
ELECTRONIC
CONFIGURATION
Ti3+, V 4+
3d1
V 3+
3d2
Cr3+, V 2+
3d3
Mn3+ Cr
M
C 2+
3d4
Fe3+, Mn2+
3d5
Fe2+
3d6
Co2+
3d7
Ni2+
3d8
Cu2+
3d9
GROUND
TERM
2D
(L=22, SS=1/2)
(L
1/2)
3F
4F
(L=3, S=3/2)
5D
6S
(L=3, S=1)
(L 2 S=2)
(L=2,
S 2)
((L=0,, S=5/2))
5D
4F
(L=3, S=3/2)
3F
2D
(L=2, S=2)
(L=3,
(L
3, SS=1)
1)
(L=2, S=1/2)
SOME OBSERVATIONS
Transition metal complexes ►
Partiall filled d
Partially
d--shell
shell,, li =2
=2,
2
degeneracy of one-electron states= 2×(2l+1)=10
dn- n electrons , d10-n- n “holes” in the fully filled d10 shell
♠
10 n shells
dn andd d10-n
h ll (complimentary
(
li
t
configurations)
fi
ti
) have
h
the same ground terms:
3d1 (one electron) and 3d9 (one hole) ►2D (L=2, S=1/2),
3d2 (two electrons) and 3d8 (two holes)►3F (L
(L=3,
3, S
S=1),
1), etc.
♠
d5- half-filled d-shell, 6S- term, L=0
(important case: total orbital angular momentum =0 ) , S=5/2.
DEGENERACY OF THE ATOMIC (IONIC)
LEVELS--REMINDER
LEVELS
Degeneracy one energy level contains
several quantum states (wave-functions):
1) 1s level (n=1, l=0) of H (hydrogen) is orbitally nondegenerate (singlet)
(singlet), this level is doubly degenerate
over spin projection :
spin “up”
up or “down”
down , ms=1/2
1/2 or -1/2;
2) 2p level (n=2, l=1) is orbitally triply degenerate
(ml = -1,
1 00, 1).
) The general multiplicity of the
degeneracy is 6 (ms=1/2 or -1/2);
In H atom there is an additional (“accidental”)
( accidental )
degeneracy. The energy levels are independent of
the q
quantum number l,, so that the energies
g
of 2s
and 2p levels are equal.
MANY--ELECTRON IONS
MANY
In many-electron atoms the value of L (total
orbital angular momentum of all electrons in the
unfilled shells) is the appropriate quantum
number that enumerates the energy levels.
The multiplicity of the orbital degeneracy is (2L+1).
The full multiplicity of the LS term is
(2L+1) (2S+1).
Example1: Ti3+ ion, 1 electron in the unfilled
d-shell( 3d1-ion ).Ground term ►2D (L=2, S=1/2)
Example2: Cr3+ ion,
ion 3 electrons in the unfilled
d-shell (3d3-ion ). Ground term ► 4F (L=2, S=3/2,
maximum
i
spin
i ffor th
three electrons).
l t
)
NOTATIONS FOR THE
WAVE--FUNCTIONS OF A FREE ION
WAVE
LSM L M S r1 , r2 ,.., rn , 1 , 2 , ,.., n
r1 , r2 ,.., rn coordinate s of the electrons
1 , 2 , ,.., n spin variables (" up" or " down" )
L quantum number of the total angular momentum
S q
quantum number of the total spin
p
M L quantum number of the projection
of the total angular momentum
M S quantum number of the projection
of the total spin
Short notation ((Dirac notation)) - q
quantum numbers :
LSM L M S
EXCERPTS FROM QUANTUM MECHANICS (REMINDER)
Physical values operators ((" cap
cap"-notat
notation for the operators):
ˆ operator of the energy, i.e. Hamiltonian
H
ˆp operator of the momentum,
momentum etc.
etc
Observable values eigen - values, eigen - functions,
ˆ E
for example : H
The wave - functions LSM L M S are the eigen
g - functions
of the following four operators : Lˆ 2 , Lˆz , Sˆ 2 , Sˆ z :
Lˆ 2 orbital angular momentum squared,
Sˆ 2 spin squared,
squared
Lˆz z - projection of the vector operator
Sˆ z - projection of the vector operator
z
ˆ,
L
Sˆ .
EIGEN--VECTORS AND EIGENEIGEN
EIGEN-FUNCTIONS:
General rules for the
angular momenta operators
of the arbitrary nature, in
particular- orbital angular
particular
momentum and spin:
Lˆ 2 LSM L M S LL 1 LSM L M S
Sˆ 2 LSM L M S S S 1 LSM L M S
Lˆz LSM L M S M L LSM L M S
Sˆ z LSM L M S M S LSM L M S
Example :
T
Term
F L 3, S
4
3
2
Eigen - functions (labels) :
3, 32 , M L M S
Lˆ 2 3, 32 , M L M S 3 4 3, 32 , M L M S
Sˆ 2 3, 32 , M L M S 32 52 3, 32 , M L M S
Lˆz 3, 32 , M L M S M L 3, 32 , M L M S
M L 3, 2, 1, 0, 1, 2, 3
Sˆ z 3, 32 , M L M S M S 3, 32 , M L M S
M S 32 , 12 , 12 , 32
ABOUT QUANTUM NUMBERS
Eigen functions
LSM L M S
Lˆ 2 LSM L M S LL 1 LSM L M S
vector L has a definite length
g LL 1
2
Sˆ 2 LSM L M S S S 1 LSM L M S
vector S has a definite length S S 1
2
LˆZ LSM L M S M L LSM L M S
Precession L around Z axis : LˆZ M L ,
LˆX LˆY 0
Sˆ Z LSM L M S M L LSM L M S
Precession S around Z axis : Sˆ Z M L ,
Sˆ X Sˆ Y 0
MAGNETIC FIELD CREATED BY AN ORBITAL
MOTION
H
Electronic orbit reminds earth orbit ,
orbital motion is equivalent to a circular electric current
that produces a magnetic field that is perpendicular to the plane
CLASSICAL ESTIMATION
Magnetic field H created by the moving
electron: proportional to the orbital angular
momentum
t
L.
Energy of interaction spin-magnetic field
E =-μS H
HL μS S
HL,
Energy of spin orbital coupling=
coupling
const LS ,
Hamiltonian of spin orbital coupling=
coupling
ˆ Sˆ ,
const L
ˆ and
L
d Sˆ are operators
SPIN--ORBITAL INTERACTION
SPIN
Interaction of the spin magnetic moment with the magnetic
field created by the orbital motion (current) of the electron
H Magnetic field created by
the orbital motion
Orbital motion
spin
Operator of spin - orbital interactio n can presented as :
ˆ Sˆ
VˆSO L
parameter of spin - orbital interactio n,
ˆ Sˆ scalar product of vector operators L
ˆ and Sˆ
L
VˆSO Lˆx Sˆ x Lˆ y Sˆ y Lˆz Sˆ z
SPIN--ORBITAL SPLITTING -QUALITATIVELY
SPIN
H
spin
spin
H
Energy of the system does depend on the
mutual orientation of the full spin
p and
magnetic field created by the orbital motion
of the unpaired
p
electron ((electrons).
)
PARAMETERS OF SPINSPIN-ORBIT COUPLING FOR THE
GROUND TERMS OF SOME TRANSITION METAL IONS
Ion
Configuration
Term
λ,cm-1
Ti3+
3d1
2D
154
V 3+
3d2
3F
104
V 2+
3d3
4F
55
Cr3+
3d3
4F
87
Mn3+
3d4
5D
85
Fe33+
3d5
6S
0
Fe2+
3d6
5D
-102
Co2+
C
3d7
4F
-180
180
Ni2+
3d8
3F
-335
Cu2+
3d9
2D
-829
COMMENT
• Spin-orbital
p
interaction is p
positive for d1,,d2 ,
d3 , d4 ions.
• Spin-orbital
Spin orbital interaction is negative for d6,dd7,
d3 , d4 ions, for the complimentary
configurations
fi
ti
dn and
d d10-n constants
t t off spini
orbital coupling are of the opposite signs.
• Spin-orbital interaction is zero for d5 , i.e.
for 6S term - S state does not carry orbital
angular momentum.
TOTAL ANGULAR MOMENTUM
Operator of the total angular momentum :
ˆ Sˆ
Jˆ L
Vector type operator, three components operators of the projections at the axes x, y and z :
Jˆ z Lˆz Sˆ z , Jˆ y Lˆy Sˆ y , Jˆ x Lˆx Sˆ x .
Properties - common for all angular momenta operators :
Jˆ 2 J , M J J 1 J , M ,
J
J
Jˆ z J , M J M J J , M J
M J J , J 1, ... , J 1, J 2 J 1 values
J , M J Dirac' s notation for the atomic wave - function
with a definite total angular
g
momenum J and p
projection
j
MJ,
i.e. quantum states with coupled spin and orbital angular momentum
TOTAL ANGULAR MOMENTUMMOMENTUM-Wave-Functions
Atomic term ► definite L and S
(Russel-Saunders coupling).
All
Allowed
d values
l
off J:
J
J = L+S,, L+S-1,, …,, | L-S |
| L-S |= L-S if L>S and | L-S |= S-L if S>L
Example: term 3F ▶ S=1, L=3 ▶ J=4, 3, 2
Labeling of the atomic wave-functions:
wave functions:
LS J MJ
Important:
this state with a definite J and MJ is a state with the definite
L and S but not (!) with the definite projections ML and MS
LABELS--Q
LABELS
QUANTUM NUMBERS
Total orbital
angular momentum
Total spin
p
angular momentum
LS J MJ
Total
angular momentum
Projection of the total
angular momentum
CLARIFICATION
• What does it mean: “definite
definite J and MJ” ?
This means that the wave-functions are the eigen-functions
of the operators Jˆ 2 and Jˆ z :
Jˆ 2 L S J M J J J 1 L S J M J ,
Jˆ z L S J M J M J L S J M J
• What does it mean: definite L and S but not (!)
definite projections ML and MS
This means that the wave
wave-functions
functions are the eigen
eigen-functions
functions
of the operators Lˆ2 and Sˆ 2 :
Lˆ 2 L S J M LL 1 L S J M ,
J
J
Sˆ 2 L S J M J S S 1 L S J M J
but not the eigen - functions of Lˆz and Sˆ z
CLASSICAL PICTURE OF COUPLING OF SPIN
AND ORBITAL ANGULAR MOMENTA
J
L
S
αS αL
X
Y
Vectors L and S precess at the conical
surfaces around vector J,
J so that the
vector sum is L+ S = J.
Because of the rapid precession of L
and S about the direction J it may be
said that mean projections of these
vectors
t
onto
t the
th plane
l
XY are zero. The
Th
length of L and the length of S remain
constant [L(L+1)]1/2 and [S(S+1)]1/2. The
constant,
angles: αL (between L and J)
and αS (between S and J) →
allowed values of J (general quantummechanical rule of momenta addition):
J = L+S,
L+S L+S-1,
L+S 1 …, | L-S
LS|
VECTOR MODEL FOR THE ANGULAR
MOMENTA IN QUANTUM MECHANICS
Z
Vector J is in a precession
about arbitrary direction Z at
the conical surface, so that
the mean values of the
projections of J onto the
plane p
p
perpendicular
p
to Z
axis are zero (JX , JY).
“Good” quantum numbers:
J and MJ
JZ
J
θ
JY
JX
X
φ
Y
J J J 1
cos
MJ
J J 1
SPINSPIN
-ORBIT COUPLINGCOUPLING-CLASSICAL ILLUSTRATION
Z
L
Vector model:
JZ
“Good”
quantum
numbers:
b
S
LS J M J
Y
X
Vector J is in a precession around Z-axis and at the same time L
and S precess around J. Length of |L| and length of |S| have definite
values but not their projections MS and ML on Z axis.
axis Vector J has a
definite length and projection MJ but mean <JX > and <JY> vanish.
SPIN--ORBIT SPLITTING
SPIN
SLJ- multiplets
p
ˆJ 2 L
ˆ Sˆ 2 Lˆ 2 Sˆ 2 2 L
ˆ Sˆ
ˆ Sˆ 1 Jˆ 2 Lˆ 2 Sˆ 2
L
2
Operator of spin orbital interactio n :
ˆ Sˆ 1 Jˆ 2 Lˆ 2 Sˆ 2
Vˆ L
SO
2
Eigen values E J can be found from the Eq. :
Vˆ L S J M E L S J M
SO
J
J
J
Vˆ SO L S J M J 12 J J 1 L L 1 S S 1 L S J M J
Enegies of the multiplets - energy as a function of J
( definite L and S ) :
EJ
2
J J 1 L L 1 S S 1
MULTIPLETS--TERMS OF
MULTIPLETS
λ>0
2
d
3F
3λ
J=4, 3F4
λ<0
-4λ J=2, 3
F2
3|λ|
4|λ|
-λ
3|λ|
d8
d2 AND d8 IONS
-λ J=3, 3
F3
3F
J=3, 3F3
-4λ J=2, 3
F2
Spectroscopic notations: multiplets 3FJ
4|λ|
3λ
J=4, 3F4
► 3F2 , 3F3, 3F4
Rule for the intervals in the multiplet structure for LS - term :
J J 1 J 1 J
2
E J E J 1 J Lande' s rule
E J E J 1
Rare--Earth IonsRare
Ions-Strong SpinSpin-Orbit Coupling
MAGNETIC MOMENT OPERATOR
Each electron in the ion or atom has
orbital angular momentum and spin.
V t operator
Vector
t off the
th orbital
bit l magnetic
ti momentt :
ˆ ˆl μˆ β lˆ , μˆ β lˆ , μˆ β lˆ
μ
l
x
x
y
y
z
z
e
Borh magneton (or B )
2mc
Spin magnetic moment :
ˆ S g e ˆs x g e β s x , y g e β s y , z g e β s z
μ
g e factor Lande, or g - factor for a free electron : g e 2.0023
ˆ ˆl , μ
ˆ 2 ˆs
μ
l
S
ˆ ˆl 2ˆs
Total magnetic moment (one - electron ion) : μ
Total magnetic moment of a many - electron ion :
ˆ 2 Sˆ , L
ˆ ˆl , Sˆ ˆs
ˆ L
μ
i
i
i
i
VECTOR MODEL FOR THE COUPLING OF THE
ANGULAR MOMENTA IN QUANTUM MECHANICS
z
Vectors L and S (of a given length)
precess around vector J at the conical
surfaces, so that the mean values of
the projections of L and S at the plane
L perpendicular to axis of J are zero
((Lx,,Ly and Sx,,Sy)). At the same time
projection of L and S at the axis of J
are non-zero and Jz=Lz+ Sz.
J
Lz
Sz
S
θ
L S cos 12 J J 1 LL 1 S S 1
y
x
J J 1 LL 1 S S 1
cos
LL 1 S S 1
Selected values for θ(J ) according to: J=L+S, L+S-1,….,|L-S|
ZEEMAN SPLITTING FOR LSJ TERMS
TERMS-- VECTOR MODEL
Let us define vector : M L 2 S attention : factor 2 g e - factor !!!
Vector M is not co - directional with the angular momentum J L S
owing to factor 2 ( g factor of the electron).
electron)
B
2S
M
C
S α
S
A
J
αL
αS- angle
l
between S and J
αL- angle
between L and J
L
Because of the rapid precession of M around the direction of
J, it may be assumed that the component BC of M averages
outt to
t zero in
i any finite
fi it time,
ti
such
h that
th t only
l the
th componentt AC
of M along J needs to be considered.
CLASSICAL VECTOR MODEL
MODEL-PROJECTIONS L AND S
S 2 J 2 L2 2 LJ cos L
L2 J 2 S 2 2 SJ cos S
Th components
The
t L cos L and
d S cos S off L and
d S along
l
J
may be expressed as :
J
L 2J
L cos L J 2 L2 S 2 2 J
S cos S
2
S2
2
These components are the projection s of the vectors L and S
onto the direction of the vector J , we assume that this is Z - axis.
axis
In fact, there is no special directions for a spherically
symmetric systems, like free atoms and ions
ZEEMAN PERTURBATION FOR A LSJ-TERM
Zeeman interactio n :
ˆ 2 Sˆ H
Hˆ μ H L
Z
g - factor for the orbital contributi on : g orb g L 1
g - factor
f t for
f the
th spin
i contributi
t ib ti on : g e g S 2
The Zeeman energy may then be written as :
EZ L cos L 2 S cos S H
Attention : factor g 2 in " spin part
part" !!!
Note : we are dealing with the classical vectors L , S and J .
3 2 S
EZ J 2 L2 S 2 2 J 2 J 2 S 2 L2 2 J H
2
L2 2 J 2 J H
ZEEMAN HAMILTONIAN FOR A
LSJ-TERM
Energy as a function of the vectors S , L and J :
E Z 3 2 S 2 L2 2 J 2 J H
This is a classical ( but not a quantum - mechanical !!! ) expression ,
all vectors are classical values but not operators.
operators
Quantum - mechanical expression
according to the rule of quantum mechanics,
mechanics
classical values should be substitute d by their operators :
ˆ 2 , J 2 Jˆ 2 , J Jˆ .
E Z Hˆ Z , S 2 Sˆ 2 , L2 L
In this way one obtains the Hamiltonia n
instead of classical energy :
ˆ 2 2 Jˆ 2 Jˆ H
Hˆ 3 2 Sˆ 2 L
Z
Finally, we have to find the Zeeman energy levels.
ZEEMAN LEVELS FOR A
LSJ-TERM
ˆ 2 2Jˆ 2 Jˆ H
Hˆ Z 3 2 Sˆ 2 L
Z axis can be chosen along the direction of the magnetic field H0 :
Zˆ 2 2 Jˆ 2 Jˆ H .
Hˆ 3 2 Sˆ 2 L
Z
Z
0
values)
Th energy levels
The
l
l (mean
(
l
)
E
L S J M Hˆ L S JM
JM J
J
Z
J
ˆ 2 are S S 1 and LL 1,
The eigen - values of Sˆ 2 and L
the eigen - values of Jˆ 2 and Jˆ are J J 1 and M .
Z
Taking this into account one can find :
E JM J g J M J H0
3 S S 1 LL 1
Notation : g J
2
2 J J 1
J
g-FACTORS FOR LSJ
LSJ--TERMS
The energy sublevels are enumerated by the quantum
numbers MJ (projection of the full angular momentum) and the
energy
gy splitting
p
g depends
p
on the field.
The value gJ is the g- factor for the LSJ-term,
g- factor for the LSJ-term is the function of L, S and J:
3 S S 1 LL 1
gJ
2
2 J J 1
Limiting cases:
• pure spin state – L=0 and J=S (orbital angular
momentum=0):
gJ= gS=2
• pure orbital state – S=0 and J=L (spin angular
momentum=0):
gJ= gL=1
gJ -SHARP
SHARP DISTINCTION FROM ge !
Rare-earth ions-strong spin-orbit coupling:
1
5
4 f , CeIII , ground term F5 2 S , L 3, J
2
2
1 3
3 4
3 2 2
6
g5 2
5 7
2
7
2
2 2
4 f 2 , Pr III , ground term 3H 4 S 1, L 5, J 4
1
2
4
g4
5
EPR of LSJ STATES
MS 1 2
g Hres
S 1 2
Hres resonance field
g
Free electron :
g e 2
Hres
ge
Hres
M S 1 2
MJ 5 2
MJ 3 2
J 5 2
2
F5 2
MJ 1 2
M J 1 2
M J 3 2
M J 5 2
H
Rare earth ion
(spin coupled to orbital angular momentum)
g J 2, g J 2
Hres
gJ
The main result :
EPR line is shifted
in the region of a strong field
MAGNETIC SUSCEPTIBILITY FOR
LSJ--TERMS
LSJ
The derivation of the magnetic susceptibility for LSJ term is
rigorously parallel to the derivation in the case of pure spin systems.
The final result can be obtained by substitution: S→J, gS→gJ
Magnetic susceptibility for a LSJ term
term:
Ng J2 2
J J 1
3kT
3kT
CJ
with C J
This leads to the Curie low:
T
Magnetization:
3k
3k
H
M Ng J J BJ y ,
Ng 2J 2
J J 1
gJ J
BJ y Brillouin function with y
kT
Important: the results are valid for a well isolated LSJ term
VALUES OF gJ AND χT FOR RARE
RARE--EARTH IONS
Ground terms SLJ for 4f n ions- see previous slide
O.Kahn, “Molecular magnetism”
Rare--Earth IonsRare
Ions-Strong SpinSpin-Orbit Coupling
MAGNETISM OF RARERARE-EARTH IONS - SOME
EXAMPLES (Gd(III) and Eu(II))
Rare earth ions contains partially filled 4 f shell ( 4 f 0 - 4 f 14 ).
Gd III 4 f 7 , EuII 4 f 7 half filled 4 f shell
Ground term :
8
S7 2 J 7 2 , L 0, S 7 2 and J S
Two main features of 4 f 7 ions : Gd III and EuII
1)Thi is
1)This
i a particular
ti l case when
h the
th orbital
bit l contributi
t ib tion to
t the
th magnetic
ti
characteristics vanishes.8 S7 2 is equivalent to a pure spin state with S 7 2
2) Excited states are very high in energy , E 6 P7 2 E 8 S7 2 30,000cm1
that exceeds considerably kT at all reasonable temperatures.
Thi means that
This
h only
l the
h ground
d term is
i thermally
h
ll populated
l d.
Since L 0, here is no spin - orbit coupling.
The magnetic susceptibility is perfectly isotropic,
isotropic
Curie low is valid for S 7 2 spin level.
MAGNETISM OF RARE
RARE--EARTH IONS SOME EXAMPLES (Sm(II)
(Sm(II) and Eu(III)
Eu(III)))
The main feature of these ions: the dependence χT vs. T does not follow
th C
the
Curie
i llow as one can expectt ffor a LSJ ground
d state:
t t
7F
0
(L=S=3)
In fact
fact, the situation is different due to the presence of
thermally populated excited states (J=1, 2, 3, 4, 5, 6):
7F , 7F , 7F , 7F , 7F , 7F
1
2
3
4
5
6
The energy levels are given by:
E J
J J 1
2
where
h
the
h energy off the
h 7F0 ground
d state is
i taken
k as an origin.
i i
Eu III and SmII special cases, excited states are close to
the ground one 300cm 1
J
6
5
4
3
2
1
0
Energy
pattern
Magnetic susceptibility should be averaged
taking into account thermal (Boltzman ) population
of the excited levels :
T
6
pJ J
J 0
where pJ is the Boltzman factor -probability of the
population of the level with the energy EJ :
pJ Z 1 exp EJ kT
J susceptibility of a quantum state with a given J :
2 2
Ng
J J J J 1
3kT
Partition function includes summation over all J 0,...,6 :
Z
6
exp EJ
kT
J 0
Energy levels EJ :
E0 0, E1 , E2 3 , E3 6 , E4 10 , E5 15 , E6 21
THE MAGNETIC SUSCEPTIBILITY
The thermally averaged susceptibi lity :
6
T
k
2 J 1 J exp J J 1 2kT
J 0
6
2 J 1exp J J 1 2kT
J 0
The factor
2 J 1 multiplici
p tyy of the degeneracy
g
y of the
J level. This factor is taken into account in the
Boltzman factor and in the summation in the p
partition function
3 S S 1 LL 1
gJ
2
2 J J 1
3
All g - factors for the excited states are equall to .
2
FINAL RESULT FOR MOLAR
χ(T)
Molar χ(T) for Sm(II) and Eu(III) ions:
6
3 N
T
4kT
2
J J 1 2 J 1exp J J 1 kT
J 0
6
2 J 1expp J J 1 kT
J 0
where all g J are replaced by 3 2 and J are substitute d.
Note: this expression is strictly valid for a free ion only.
only
Influence of surrounding in crystal and complexesa separate question. Crystal field splits (in general)
J-multiplets
p
and affects magnetic
g
p
properties.
p
χT versus
THE NUMERICAL RESULT:
kT/λ PLOT FOR AN Eu(II) COMPOUND
χT is temperature dependent, i.e. does not follow the Curie
low. At T=0 the product χT→0 due to the fact that χ(J=0)=0.
χT increases with the increase of temperature due to thermal
pop lation of the states with
population
ith high J that contribute
contrib te to the
susceptibility.
Chapter IV
Effects of crystal field.
Group theoretical introduction.
Group-theoretical
introduction
Ground terms of the transition metal
ions in the crystal fields
fields. Anisotropy of
the g-factor. Zero-field splitting:
qualitative and quantitative
approaches. Covalence and orbital
reduction EPR of the metal ions
reduction.
in complexes.
CRYSTAL FIELDFIELD-THE MAIN PROBLEM
Me
Free metal ion
Men+ in a LS or
LSJ state
Me
Coordinated ion
M (li
Me(ligand)
d)6 -ligand
li
d
surrounding in a complex
compound or in a crystal
The main questionquestion-how the surrounding affects the energy
l
levels
l and
d th
the magnetic
ti properties
ti
Hans Bethe ,
Nobel winner,1967
German-born
G
b
A
American
i
theoretical physicist who helped
to shape classical physics into
quantum physics and increased
the understanding of the atomic
processes responsible
p
p
for the
properties of matter and of the
forces governing the structures of
atomic nuclei.
nuclei He received the
Nobel Prize for Physics in 1967
for his work on the production of
energy in stars
stars. Moreover
Moreover, he was
a leader in emphasizing the social
responsibility of science.
J.H. Van Vleck
American physicist and mathematician who shared the Nobel Prize for
Physics in 1977 with Philip W. Anderson and Sir Nevill F. Mott. The prize
honoured Van Vleck's contributions to the understanding of the behaviour of
electrons in magnetic, noncrystalline solid materials.
Van Vleck developed during the early 1930s the first fully articulated
quantum mechanical
h i l theory
h
off magnetism.
i
L
Later
h was a chief
he
hi f architect
hi
off
the ligand field theory of molecular bonding. He contributed also to studies
of the spectra of free molecules, of paramagnetic relaxation, and other
topics. His publications include Quantum Principles and Line Spectra (1926)
and the Theory of Electric and Magnetic Susceptibilities (1932).
SPLITTING OF THE ATOMIC LEVELS
IN CRYSTAL FIELDS
Each atomic (ionic) level with a given L or J is
split in a crystal field.
STATEMENTS AND RULES DERIVED FROM THE
BACKGROUND OF THE GROUP THEORY:
THEORY
1) (2L+1) wave
wave-functions
functions belonging to the atomic level with
a given L ( LS- term) form the basis of a degenerate
irreducible representation of the full spherical symmetry
group R3.
2) This representation is referred to as D(L), basis (wave
(wavefunctions) is formed by (2L+1) wave-functions of the type
of YLM ((spherical
p
functions),
),
M=-L,-L+1,…, L-1, L, ► (2L+1) - values.
3) Point symmetry of the atom (ion) in a crystal or in a
ligand surrounding in a complex compound is lower than
the spherical one (R3). Under this condition the
representations D(L) become reducible. Each reducible
representation
p
can be decomposed
p
into irreducible
representations (in the point symmetry group)
possessing low dimensions.
4) Each irreducible representation (in R3 or in the crystal
symmetry group) corresponds to an one energy level.
5) The
Th physical
h i l consequence off these
th
mathematical
th
ti l
conclusions is that each atomic level becomes split (in
general) when the atom (ion) is placed in the ligand
surrounding: instead of one ionic terms SL one obtains
several crystal
y
field terms ((crystal
y
field splitting).
p
g)
BOOKS ON GROUP THEORY AND CRYSTAL
FIELD THEORY
1.F.A.Cotton,
1
F A Cotton Chemical Application of Group Theory
Theory,
2nd Edition,Interscience, New York (1971).
2. B.S.Tsukerblat,, Group
p Theory
y in Chemistry
y and
Spectroscopy. A Simple Guide to Advanced Usage,
Academic Press, London, 1994.
3. Robert L.Carter, Molecular Symmetry and Group
Theory, John Wiley, 1998.
4 S
4.
S.Sugano,
S
Y
Y.Tanabe,
T
b H
H.Kamimura,
K i
M
Multiplets
lti l t off
Transition Metal Ions in Crystals, Academic Press,
New-York 1970
New-York,
1970.
5. C.L. Ballhausen, Introduction to the Ligand Field Theory
and its Applications,
pp
, Pergamon
g
Press,, Oxford,, 1963.
THE MAIN PROBLEM IN QUESTION:
HOW THE FREE ION TERMS (SL) ARE SPLIT IN A
CRYSTAL FIELD-CRYSTAL FIELDS TERMS (SΓ) AND
CRYSTAL FIELD SPLITTINGS
Irreducible representations Γ (irreps) of the point group Oh
(cubic group -octahedral
octahedral or cubic surrounding of the ion):
Even irreps : A1g, A2g - one-dimensional irrep
Eg bi dimensional irrep
bi-dimensional
T1g , T2g -tri-dimensional irreps
Odd irreps:
i
A1u , A2u - one-dimensional
di
i
l irreps
i
Eu -
bi-dimensional irrep
T1u , T2u - tri-dimensional
tri dimensional irreps
Important notation: “g”-even (gerade), ”u”-odd (ungerade)
(parity of the crystal field states
states, for the
point groups with the inversion symmetry)
STRUCTURE OF THE CYANOMETALATES
FAMILY
Metal ion
CN-group
An example of the octahedral metal complex,
Oh symmetry:
t Metal(CN)
l( )6
eg-orbitals
z
3z2-r2
zx
x
x2-y2
t2g-orbitals
yz
xy
y
Shape of d
d-orbitals
orbitals and splitting
HOW TO FIND THE SPLITTING OF SL IONIC TERMS
IN THE OCTAHEDRAL LIGAND SURROUNDING?
RESULTS for several values of L: (decomposition in oh group,
even ionic states, d-electrons, l=2 )
ANSWER: to decompose the reducible D(l) irreps into the irreducible
ones (the procedure is well known from the group theory)
symbolically:
b li ll
DL
L irreps
D 0 A1g
S A1g singlet
g
D 1 T1g
P T1g triplet
D 2 E g T2 g
D E g T2 g doublet
d bl triplet
l
D 3 A2 g T1g T2 g F A2 g T1g T2 g singlet
g two triplets
p
each irrep ◄►energy level in crystal field (crystal field splitting)
PHYSICAL PICTURE OF THE CRYSTAL FIELD
SPLITTING
Shapes of the electronic clouds:
r wave function
r spatial distributi on
2
of the electronic density
(shape of the electronic " cloud" )
Energy in the crystal field interactio n
of the electronic cloud with
the charges of the ligands
SHAPES OF THREE
p-ORBITALS
dumbbell-shaped
electronic clouds
p-ORBITALS IN THE
OCTAHEDRAL
O
AHEDRAL (Oh)
CRYSTAL FIELD
Positive
(black) and
negative
(light) petals
of the wavewavefunctions
CONCLUSION FROM THE PICTURE
•The energies of the interaction of the dumbbellshaped
h
d electronic
l t i clouds
l d off three
th
p-orbitals
bit l with
ith
the ligands of the octahedral surrounding are
equal.
l
•Three p-orbitals form triply degenerate level in the
octahedral
t h d l crystal
t l fi
field.
ld
•This is the physical sense of the group-theoretical
statement D(1)→T1u (the only triply degenerate
irrep, this means that there is one triply
degenerate level in a cubic crystal field) .p-level
remains degenerate in the cubic (octahedral
crystal surrounding.
Five d-orbitals
dxz
dxz
y
two dumbbells in
each: (yz,xz,xy)
(3z2-r2, x2-y2)
Five dd-orbitals in the octahedral field
z
z
y
y
z
x
dxz
z
y
y
x
(yz,xz,xy) T2-orbitals
(3z2-r2, x2-y2) E-orbitals
ELECTRONIC STATES (TERMS)
IN CRYSTAL FIELD – LABELS
Spin
multiplicity
2S 1
2T
Irreducible
representation
- orbital triplet, S=1/2,
2E - orbital doublet
doublet, S=1/2, etc.
etc
g
2g
IMPORTANT REMARK: PARITY RULES
1) one electron: p(parity) = (-1)l
p-electron: l=1 (odd states)
d-electron: l=2 ((even states))
f-electron: l=3 (odd states)
2) Many (n) electrons: ( but not (-1)L !!! )
dn- shells,
h ll allll li=2
2 (even
(
states
t t )
p1(odd), p2 (even), p1d1(odd), etc.
In the point symmetry groups involving inversion center:
u odd irreps , g even irreps
SPLITTING OF THE dd-LEVEL IN A CUBIC
FIELD INTO A TRIPLET AND DOUBLET
Rnl r Ylm ,
Five d-functions (angular parts): Y2,-2, Y2,-1,Y2,0,Y2,1 Y2,2
d level (l=2) ►
d-level
D(2)→T2+E (triplet +doublet)
T2(xy,
(xy xz,
xz xy) (real) and E( 3z2-rr2, x2-yy2)(real)
5-fold degenerate d-level is split into
a triplet and a doublet in a cubic crystal field
field.
Notations for the d-functions in Oh:
d yz , d xz , d xy T2 g
and d
x2 y2
,d
z2
E g
CRYSTAL FIELD SPLITTING IN THE CASE OF
ONE d-ELECTRON OR ONE HOLE
One d-electron, l = 2.
D
2
E g T2 g D E g T2 g doublet triplet
2
Eg
2
T2 g
2D
2D
10Dq
10Dq
2
T2 g
d1- electron
l t
(Ti
(T 3+)
2
Eg
d9-hole
h l (C
(Cu2+)
d 9-“hole” in the closed shell d 10
((reversed order of the levels:
in Ti3+-ground triplet, in Cu2+-ground doublet )
Physical reason:
electron-negative charge-“cloud” (repulsion
f
from
the
th ligands),
li
d ) h
hole-positive
l
iti charge-”cloud”
h
” l d”
(attraction to the ligands).
10Dq- cubic crystal field parameter =
p
g of the one-electron level ((d1) in
splitting
a cubic crystal field
CUBIC CRYSTAL FIELD PARAMETER 10Dq
D
Dq
eq r 4
5
6R0
q*
Metal
<r4>
R0
Ligand
Point
P
Pointi t-charge
h
model
d l for
f
the crystal fieldfield-ligands
are the point charges
(covalency is not taken
into account):
q* -charge of the ligands
(point charges,
R0-metal-ligand
distances in the
octahedral surrounding
10Dq- crystal field
splitting of the
one-electron d- level
<r4>-mean value of r4
for the d-electron
CRYSTAL FIELD SPLITTING : d2 and d8 IONS
IN THE OCTAHEDRAL (Oh) FIELD
d-electrons: l1= l2=2, L=3 (F-term)
3
3
T1g
A2 g
3F
10Dq
8Dq
3
T2 g
3
T2 g
3F
8D
8Dq
10D
10Dq
3
T1g
d 2 (V 3+)
3
2 electrons
A2 g
3
A2 g
d 8 ( Ni 2+)
2 “holes”
holes in d10
d 8- two “holes” in the closed shell d 10 (reversed order of the levels: in V 3+ground triplet,
triplet in Ni 22+-ground
ground singlet ) 10Dq
10Dq- cubic crystal field parameter =
splitting of the one-electron level (d1) in a cubic crystal field
Typical (experimental) values of Dq in transition
metal complexes with H2O ligands:
(Y.Tanabe, S.Sugano, J.Phys.Soc.Jap.9,766(1954))
Ion
Ti3+
V3+
Cr3+
Mn3+
Fe3+
Co3+
Dq cm-1
Dq,
2030
1860
1720
2100
1350
1920
Ion
Cr2+
Mn2+
Fe2+
Co2+
Ni2+
Cu2+
Dq, cm-1
1390
1230
1030
840
820
1220
Some conclusions:
1) crystal field splitting is of the order of 10,000-20,000cm-1(visible
region the light);
2) empirical rule: irrespective of the ligand and metal 10Dq in
the systems with divalent ions is around 10,000 cm-1 metal
ions and in those with trivalent metal ions around 20,000cm-1.
SPECTROCHEMICAL SERIES
When the metal element is fixed and the ligand is varied,
the magnitudes of 10Dq may be arranged
in the following order:
I < Br < Cl < S < F < O < C
where the elements are those in ligands attached
directly to the metal
(Tsushida’s
(Tsushida
s spectrochemical series)
When the ligand is fixed and the metal ion is varied
the magnitudes
g
of 10Dq
q mayy be arranged
g
in the following order:
Mn2+ < Ni2+ < Co2+ < Fe2+ < V2+
< Fe3+ < Cr3+ < V3+ < Co3+
< Mn4+ < Mo3+ < Rh3+ < Pd4+ < Ir3+
< Re4+ < Pt4+
TETRAGONAL Cu(II) COMPLEXES
D4h point symmetry group
Oh→D4h
Elongated
octahedron
Compressed
octahedron
Mixed-ligand MeA4B2 complexes
ENERGY LABELS AND SPLITTING
When the symmetry is lowered, according to the rules of group
theory the degenerate levels become split .
Using the characters table and the rule of the decomposition
one can find:
(Oh-group)
(D4h-group)
T2g (yz, xz, xy) → Eg (yz, xz) + B2g(xy)
Eg (z2, x2-y2) → A1g (z2) + B1g(x2-y2)
Orbital triplet is spit into the doublet and singlet, orbital
doublet is split into two singlets.
The group-theoretical results provides the labels for the levels
((irreducible representations)) and the multiplicity
y of the energy
gy
levels, but the energies of the levels in the tetragonal crystal
field should be calculated in a quantum mechanical way.
N
Nevertheless,
th l
th
the order
d off the
th split
lit llevels
l can be
b d
derived
i d ffrom
the qualitative arguments.
SPLITTING OF THE GROUND STATE OF A
Cu(II) COMPLEX IN A TERAGONAL FIELD
2
T2 g
2
Dd
2
2
9
10Dq
2
free
Cu II
E g yz , xz
2
Eg
Oh
B2 g xyy
A1 g 3 z 2 r 2
2
B1g x 2 y 2
Energy pattern
for an elongated
g
octahedrally
coordinated or
square-planar
Cu(II) complex .
D4 h
Problem:
evaluation
l ti off th
the g-factor
f t for
f the
th ground
d state
t t in
i a crystal
t l field
fi ld
GROUND STATE OF A dd-HOLE
HOLE-A QUALITATIVE ORBITAL PICTURE
Z
Compressed
octahedron
Y
X
Z
3z 2 r 2 A1g
Elongated
octahedron
2
x y
Compressed
p
conformati on :
efficient attraction of d
z2
" cloud"
to the apical
p
ligands
g
Elongated conformati on :
Y
X
Ground state of the d9
electronic shell in a
tetragonal crystal field is
defined from the condition of
th mostt efficient
the
ffi i t attraction
tt ti
of the positive charge
((“hole”)) to the ligands
g
negative charges.
2
B1g
efficient attraction of d
x2 y2
" cloud"
to the equatorial ligands
ORBITAL DIAGRAM FOR THE GROUND STATE OF
A Cu(II)
( ) COMPLEX
P E IN A TETRAGONAL
E
G
CRYSTAL FIELD
F E
E -orbitals in a
cubic field (one
half-filled orbital)
x2-y2
z2
10Dq
10D
T2 -orbitals in a
cubic field (fully
occupied)
i d)
xy
xz, yz
Next stepstep-calculation of the g-factors
ORBITAL PICTURE OF THE
MULTI--ELECTRON EXCITED STATES
MULTI
x2-y2
z2
xy
xz, yz
|x2-y2>
|z2>
unpaired electron
|
|xy>
|
|yz>
|
|xz>
Degenerate, 2E
Degenerate
MATRICES OF SPIN OPERATORS
For spin
1
2
one can define the so - called spin basis, i.e. two
spin - functions : a and and .
Matrix elements of the operators Sˆ x , Sˆ y and Sˆ z are defined within
thi basis.
this
b i This
Thi means that
th t each
h operator
t Sˆ u has
h four
f
matrix
t i elements
l
t :
| Sˆ | , | Sˆ | , | Sˆ | , | Sˆ |
u
u
u
u
S i - matrices
Spin
i
collection of the matrix elements represented as a square matrix :
| Sˆ | | Sˆ |
u
u
| Sˆu | | Sˆu |
This matrix (for each operator Sˆu ) has two rows and two columns 2x2 - matrix (second order matrix) in the two - dimensional basis a and .
The matrices of spin - operators are the following :
ˆS 1 0 1 , Sˆ 1 0 i , Sˆ 1 1 0 , i 1
x
y
z
2 1 0
2 i 0
2 0 1
To find out the meaning of these matrices one should compare
them with the general definition u x , y , z :
| Sˆ u | | Sˆ u |
| Sˆu | | Sˆu |
One can see that | Sˆ x | 0 , | Sˆ x | 1 2 , etc.
The matrices :
ˆx
0 1
1 0
ˆy
,
0 i
i
0
ˆz
,
1
0
0 1
are known as the Pauli matrices,
1
1
1
ˆ x , Sˆ y
ˆ y , Sˆ z
ˆz
Sˆ x
2
2
2
MATRICES OF THE OPERATORS Lx , Ly and LZ USING
THE dd-ORBITALS AS A BASIS SET
MATRIX ELEMENTS OF THE OPERATORS LX, LY and LZ
We shall consider an elongated
g
conformati on of a Cu(II)
( ) complex.
p
The matrix elements connecting the ground state d 2 2
x y
with the excited ones d xy , d yz and d xz are the following :
d xy | Lˆz | d
d yz | Lˆ x | d
x2 y2
x2 y2
2i
d xz | Lˆ y | d
x2 y2
i
Important remark :
the matrix elements connecting d
x2 y2
and d
z2
orbitals vanish. It can be said that the mean value of
the angular momentum operator is zero in the
cubic term E g basis : d 2 2 and d 2 .
x y
z
Physical conclusion :
cubic crystal field " kills
kills" orbital magnetic contributi on
in the ground state E g of the " hole" (the case of Cu 2 ion).
Cu2+-ION IN A MAGNETIC FIELDFIELDZEROTH
E
H ORDER
DE APPRXIMATION
X M
N
Let us evaluate g - factor for the ground state
of a d 9 ion with the ground d
x2 y2
state.
state
Two wave - functions of the ground state :
d
x2 y2
d
x2 y2
and d
x2 y2
d
x2 y2
Zeeman interaction :
ˆ 2 Sˆ H
Hˆ Z B L
ˆ H,,
Orbital
O
b ta pa
partt B L
Spin pa
Sp
partt 2 B Sˆ H
ˆ vanish :
Matrix elements of L
d
x2 y2
ˆ d
L
x2 y2
0
Result :
mean value of the orbital part of Zeeman interaction 0
Zeeman energy in the case of Z - direction of the field HZ H0 :
E d
x2 y2
2 B H0 Sˆ z d
E d
x2 y2
2 B H0 Sˆ z d
B Lˆ z 2 Sˆ z H0 d
x2 y2
d
x2 y2
x2 y2
Normalizat ion : d
x2 y2
x2 y2
d
1
2 B H0
2
B Lˆz 2 Sˆ z H0 d
d
x2 y2
x2 y2
1
2 B H0
2
x2 y2
1
This can b
Thi
be expressed
d as :
E M S 2 B H0 M S g B H0 M S
Conclusion :
g 2 pure spin value,
orbital contributi on has dissapered
due to action of the crystal field of D 4h symmetry
SOME RULES OF PERTURBATION THEORY IN QUANTUM
MECHANICS
ˆ and Vˆ :
We suppose that the Hamiltonia n consists of two parts : H
0
ˆ Hˆ Vˆ
H
0
ˆ the main part - unperturbe d Hamiltonia n.
H
0
Vˆ perturbati on operator, the perturbati on is assumed to be relatively small.
0
Let us denote the unperturbe d ground state wave function as gr
,
ˆ :
this function is the eigen - function of the unperturbe d Hamiltonia n H
0
ˆ 0 E 0 0
H
0
gr
gr
gr
We assume that we can solve this Schr鰀inge r equation
0
and so,
and,
so know this wave - function as well all the energy E gr
and
all zero - order energies of the excited states .
Let us evaluate the wave - function of the g
ground state at the first order.
0
Under the action of perturbati on the wave - function gr
proves to be modified
1
and gets a relatively small additional term (correctio n) gr
,
0
1
First order wave - function gr
gr
to be calculated .
QUANTUM - MECHANICAL RULE
FOR THE EVALUATION OF THE CORRECTION:
n | Vˆ | gr 0
1
gr 0
0 n ,
n E gr En
where n | n0 are the unperturbed wave - functions of the excited states " n" ,
n | Vˆ | gr matrix elements of the perturbation between ground and excited
0
unperturbed energy of the ground state,
states in the unperturbed basis, E gr
En0 unperturbed energy of the excited state n | .
Summation includes all excited states.
The perturbation theory gives a good result if the correction is small enough,
the condition of the applicability of perturbation theory can be expressed as follows :
n | Vˆ | gr E 0 E 0
gr
n
This means that the ground state should be well isolated from the excited ones ,
i.e. absolute values of all matrix elements n | Vˆ | gr are much smaller
0
than the energy gaps E gr
En0 .
MATRIX ELEMENTS OF SPINSPIN-ORBITAL
INTERACTION
Ground state wave - functions (including spin components ) :
d
x2 y2
and d
x2 y2
Spin orbital interaction :
Vˆ Lˆ Sˆ Lˆ Sˆ Lˆ Sˆ
SO
x x
y
y
z z
Mean value of spin - orbital interaction in the ground state 2B1g is zero,
due to the fact that all matrix elements of Lˆu vanish :
d
x2 y2
Lˆx Sˆ x d
x2 y2
d
d
x2 y2
Lˆz Sˆ z d
x2 y2
d
x2 y2
Lˆx d
x2 y2
Sˆ x 0,
x2 y2
Lˆz d
x2 y2
Sˆ z 0,
etc.
For this reason the mean value of the
orbital part of the Zeeman interaction vanishes.
MIXING WITH THE EXCITED STATED
Spin - orbital interactio n does not affect the ground state in
zeroth order approximat ion.
Let us evaluate the wave - function of the ground state at the first order.
Zeroth order functions : d
x2 y2
and d
x2 y2
,
Pertutbati on operator : VˆSO ,
Zeroth order energies
g
- energies
g
in the tetragonal
g
crystal
y
field,,
1 and 2
Quantum - mechanical rule for the evaluation
the required matrix elements of Vˆ can be calclated with the help
SO
of matrices of Lˆu and Sˆ u , all matrix elements of Lˆu and Sˆ u can be extracted
from these matrices, for example :
d yz | Lˆ x Sˆ x | d
x2 y
ˆ
2 d yz | Lx | d
ˆ | i 1 i
|
S
x
x2 y2
2
2
GROUND STATE OF Cu(II) COMPLEX TO FIRST ORDER
The rule and some steps are already given:
The first order wave functions for the ground term
in an elongated or square planar Cu(II) complex
are the following:
d
x2 y2
Energy
pattern
2
d xy | Lˆ z Sˆ z | d 2 2 1 d xy
x y
d yyz | Lˆx Sˆ x | d 2 2 2 d yyz
x y
d xz | Lˆy Sˆ y | d 2 2 2 d xz
x y
d 2 2 d xy | Lˆz Sˆ z | d 2 2 1 d xy
x y
x y
d yz | Lˆ x Sˆ x | d 2 2 2 d yz
x y
d xz | Lˆ y Sˆ y | d 2 2 2 d xz
x y
10Dq
E g yz , xz
2
B2 g xy
2
A1 g 3 z 2 r 2
2
B1g x 2 y 2
Δ2
Δ1
D4 h
Energy gaps
Δ1 and Δ2
FINAL FORM OF THE WAVE FUNCTIONS
After calculatio n and substituti on of all matrix elements one finally finds
i
i
d xz
d
d
xy
2 yz
x2 y2
1
2
2 2
i
i
d yz
d xz
d 2 2 d xy
d
1
2 2
2 2
The function shows that the ion exists not only in the state d
x y
x2 y2
but also, in part, in the excited states d xy , d yz and d xz
(principle of superposit ion in quantum mechanics) ;
theses parts are small, in fact :
1
1,
2
1
A similar conclusion can be drown with regard to .
This is a condition for the applicabil ity of the pertubatio n theory.
REMARK REGARDING THE WAVE
WAVE-FUNCTIONS
Wh do
Why
d we need
d the
th wave - functions
f
ti
t the
to
th first
fi t order?
d ?
Orbtal contribution within the basis set of
zeroth order appximatio n is strictly zero,
Orbtal contribution within the basis set to a
first order approximat ion is non - zero.
I fact,
In
f t spin
i - orbital
bit l interactio
i t
ti n gives
i
smallll addition
dditi
of the orbital magnetic moment to the ground state
so that the ground state possess now both : spin and orbital
contributions to the magnetic
g
moment .
HOW TO EVALUATE g
g--FACTORS?
I order
In
d to
t calculate
l l t the
th g - factors
f t
we mustt
calculate the Zeeman splitting
with
ith the
th use off the
th first
fi t order
d ground
d state
t t
wave functions and .
ˆ 2 Sˆ H
Zeman operator : Hˆ Z B L
Operators Sˆ and Lˆ have only diagonal matrix elements,
z
z
operators Sˆ x , Sˆ y and Lˆx , Lˆy have only off - diagonal matrix elements
(see matrices of these operators).
Non - zero matrix elements (with the use matrices of Lˆi and Sˆi ) :
Lˆ z 2 Sˆ z , Lˆ x 2Sˆ x
and
d Lˆ y 2Sˆ y
,
so the matrix of the Zeman operator within the basis set ,
contains diagonal and off - diagonal matrix elements.
ANISOTROPY OF THE gg-FACTOR - SEQULAR
EQUATION
The aim : to find eigen - values of the Zeeman operator as functions of the
applied magnetic field, this is a way to determine g - factors.
Accordingly to the rule of quantum mechanics, one should build the
matrix of the perturbation and then to diagonalize this matrix.
ˆ general form :
2x2 - matrix of H
Z
ˆ
H
Z
ˆ
H
Z
ˆ
H
Z
ˆ
H
Z
To find the eigen
g - values ( Zeeman levels, E ) one should solve
the following equation ( so - called secular equation, determinan t, 0) :
Hˆ Z E
Hˆ Z
Hˆ Z
0
ˆ
H Z E
This is a quadratic algebraic equation that can be solved (relative E )
always providing arbitrary direction of the external magnetic field.
ANISOTROPY OF THE g
g--FACTOR – SECULAR EQUATIONS
FOR TWO PRINCIPAL DIRECTIONS
We shall consider this equation in the cases of two orientation
of the magnetic field along principal directions - parallel to C4 axis
and in the equatorial plane C4 , let say, parallel to X axis.
The case of " parallel" field H || C4 :
B LˆZ 2 Sˆ Z HZ E
0
0
0
ˆ
ˆ
B LZ 2S Z HZ E
The case of " perpendicu lar
lar" field H C4 , H || X :
E
B LˆX 2Sˆ X H X
B Lˆ X 2Sˆ X H X
E
0
All matrix elements are to be substitute d from the matrices of Lˆi and Sˆi
EXPRESSIONS FOR THE gg-FACTORS
From these secular equations one can find the energy levels as the
functions of the external magnetic field,
then we can find out the g - factors.
THE FINAL RESULT :
g Z ge
8
,
1
g X gY g e
2
2
Generally accepted notations :
g Z g|| , g X gY g
8λ
2λ
and
Δ1
Δ2
anisotropic contributions,
i.e contributions that depend on the DIRECTION of the applied field.
These important values depend on the spin - orbit coupling λ (including sign ! )
and tetragonal crystal field splitting parameters Δ1 and Δ2
CONCLUSION--EFFECT OF CRYSTAL FIELD
CONCLUSION
1)) g factors are diffrent from g e 2 value for a free electron;;
2) g factors do depend on the orientation of an external magnetic field respectively
crystal (molecular ) axes, in fact : g|| g .
Thus, g factors of the metal ions in a crystal field become anisotropic .
3) Resonance conditions in EPR become dependent
off the
th orientatio
i t ti n off the
th crystal
t l axes respective
ti ly
l the
th magnetic
ti field
fi ld H.
3) In the case of D4 h symmetry the g factors are axially symmetric :
g X gY g Z
The magnetic splitting is independent of the orientation of the field in the plane C4
and depends only on the angle between Z - axis and vector of the field H.
4) In the In the case of D4 h symmetry and ground d
x2 y2
orbital the condition
g|| g
is always valid. In fact,
8
2
8
2
and
are p
positive 0 for d 9 ion and
1
2
1
2
Typical values for Cu(II) complexes : g|| 2.20, g 2.08
1 2
ADDITIONAL REMARKS ABOUT THE
ORBITAL CONTRIBUTION
The main physical question:
why g
g--factors in a crystal field are different from ge=2 and anisotropic ?
• Crystal field (electric field of ligands-charges) does not interact
directly with the spin (magnetic moment).
moment)
• Crystal field affects only the orbital motion of the electron in the
unfilled electronic shell.
• Orbital
O bit l motion
ti iinteracts
t
t with
ith the
th spin
i (spin-orbital
( i
bit l coupling)
li ) and
d th
thus ,
indirectly, spin (through the orbital motion) interacts with the crystal
field and through
g this interaction spin
p “feels” axes of the crystal
y
field.
• How this appears in the quantum-mechanical approach? The wavefunctions (to the first order approximation) contain terms proportional
to λ , these terms are small but they give rise the anisotropic
contributions to g-factors. In fact, without these corrections (“admixture”
of the excited states)) to the wave-functions all matrix elements of the
operator L in the ground state would be strictly zero.
ANISOTROPY OF THE MAGNETIC SUSCEPTIBILITY
The case of isotropic systems ( free ions) :
Ng 2 β 2
χ
S S 1
3kT
1
Anisotropi c systems (axial symmetry, S ) :
2
Ng||2 B2
H || C4 χ||
4kT
Ng 2 B2
H C4 χ
4 kT
Powder (polycryst alline) samples - in the analysis of the magnetic data
the averaged values of the g factors are usually used :
1 2 2 2
g|| g
3
3
1
Tri - axial symmery : g 2 g x2 g 2y g z2
3
Axial symmery : g 2
SCHEME OF THE EPR TRANSITIONS IN TWO MAIN
ORIENTATIONS OF THE APPLIED FIELD
g Hres , Hres resonance field,
+
S
1
2
frequency of the oscillating field
" Parallel" field
g|| g e
S
1
2
+
H||
H H
8
1
Hres,||
" Perpendicular" field
2
g ge
2
Hres,
g||
g
g|| g , H H||
C
Conclusion:
l i
position
iti off th
the EPR line
li d
does d
depend
d on th
the
direction of the field respectively of crystal axes.
ANGULAR DEPENDENCE OF THE g
g--FACTOR IN A
TETRAGONAL
E
G N L SYSTEM
Y EM
Arbitrary orientation of the magnetic field
B LˆZ 2Sˆ Z HZ E
B LˆX 2Sˆ X H X
HX H sin cos ,
B LˆX 2Sˆ X H X
B LˆZ 2Sˆ Z HZ E
HY H sin sin , HZ H cos
g factor for an arbitrary direction of the magnetic field
((angular
g
dependence
p
of g factor),
), result :
g 2 g||2 cos 2 g 2 sin 2
Z
H
φ
X
Note : Symmetry of the g - factor is higher
than the point symmetry group.
Y
In fact p
point symmetry
y
y is D 4h ,,meanwhile
g - factor is axially symmetric.
This is valid for all point groups
involving axes Cn with n 3,
C3v , C4v , D4 h , etc.
0
POWDER SAMPLESAMPLE-THE PROBLEM
Powder sample,
or a frozen solution containing paramgneti c centers
( l
(molecules
l , metal
t l complexes)
l
) in
i a non - magnetic
ti substance
b t
fully disordered (random ) orientatio ns of the magnetic axes.
The main question
question-is
is it possible to extract information
about the magnetic ions from the EPR spectra if the
principal directions of the g-factors are fully disordered?
disordered
SHAPE OF THE LINE IN A POWDER SAMPLE
1
S
2
+
Z
θ=0
θ
Right
bound
θ=π/2
Left
bound
H|| H H
EPR line of a
powder sample
has left and right
bounds, i.e.
values of the
resonance field
corresponding to
principal directions
of the applied field
respectively ZZ
axes of randomly
oriented
molecules.
l
l
Absorption occurs in a restricted range of the field.
EPR OF A POWDER (POLYCRYSTALLINE)
SAMPLE-- SHAPE OF THE EPR LINE
SAMPLE
Calculated
shapes
g
g||
g||
gZ
g
gY
gX
Axial symmetry,
shape of the line - sharp peak
at the right bound
(bounds are smoothed)
Axial symmetry,
derivative of the line
Tri-axial symmetry,
symmetry
derivative of the line
WIDTH OF THE LINE IN A POWDER SAMPLE
g ge
2
2
8
g|| g e
1
Hres,
Hres,||
right bound
bo nd of the line
g
left bound of the line.
g||
Width of the line :
Hres, Hres,||
1
1
g g||
g g||
Broadening of the EPR line effect of the magnetic anisotropy.
TRI--AXIAL SYMMETRY
TRI
SYMMETRY--SOME RESULTS
In the case of triaxial symmetry g - factor has three different components :
g X gY g Z
g 2 g x2 cos 2 x g 2y cos 2 y g z2 cos 2 z
x , y ,z angles between vector H and axes X,Y,Z.
Point symmetry groups containing C2 axes : C2v
2 , D2h ,etc .
C2Z
B
C
A
C2Y
C2X
Application of the EPR technique to a single
crystal as well as to a powder sample
COVALENCY AND EFFECT OF ORBITAL
REDUCTION
Crystal field theory considers ligands as the point charges. Really they
have their own electronic structure and ligand
g
orbitals. More careful
consideration takes into account covalence, metal orbitals and ligand
orbitals are mixed-molecular orbital approach.
X
Y
Z
Z
Atomic d-orbitals and p-orbitals of the ligands participating in the formation
of the molecular orbitals |xz> and |yz> in an octahedral complex
ORBITAL REDUCTION FACTOR
Molecular orbitals - linear combination of the atomic orbitals
possessing a definite symmetry.
XZ c1d xz c2 L xz
YZ c1d yyz c2 L yz
L xz , L yz linear combinations of the atomic p - orbitals
that are transformed like xz and yyz under the g
group
p operations
p
.
c1 and c2 numerical coefficients that are found from the computation
within the molecular orbital approach,
pp
,
c12 2c1c2 ML c22 1
ion
c12 the most part of the electronic density on the metal ion,
c22 the most part of the electronic density on the ligands
ML metal - ligand overlap integral
REDUCTION FACTOR
Calculation of the matrix elements
of the orbital angular momentum shows
that this physical value is reduced ,
ˆ should be substituted by a new effective value :
the operator L
ˆ kL
ˆ , with k 1
L
Very roughly : k c12 2c1 c2 ML ,
ML d xy L xy ,etc metal - ligand overlap,
overlap
the part of the electronic density localized at
the metal ions contribute s to the algular moment.
This is known as : the effect of the reduction
of the orbital angular momentum by covalence.
PHYSICAL CONSEQUENCES
OF THE REDUCTION EFFECT
Spin - orbital interactio n is reduced :
ˆ Sˆ , k
Hˆ kL
SO
It can be said that only the metal ion contribute s to .
Orbital part of the Zeeman interactio n is reduced,
Zeeman interactio n in a covalent complex should be witten as :
ˆ 2 Sˆ H ,
Hˆ Z B kL
ˆ L
ˆk
L
Anisotropic parts of the g - factors are reduced :
8k
g Z ge
,
1
2k
g X gY g e
, k
2
For the transition metal ions the reduction factor can be estimated as :
0.6 k 0.9
A more precise consideration shows that the reduction factors
are anisotropic.
ZERO-FIELD SPLITTING
ZEROSPLITTING-GROUND STATE OF Ni2+ COMPLEXES
x2-yy2
3
T1g
3F
10Dq
z2
3
T2 g
8D
8Dq
3
A2 g
d 8 ( Ni 2+)
2 “holes”
holes in d10
t 26 e 2
10Dq
xy
xz, yz
Octahedral crystal
y
field, ground
g
state 3A2g –orbital
singlet (orbitally non-degenerate) and full spin S=1 ( spin triplet)
EFFECT OF THE CUBIC CRYSTAL FIELDFIELD-MORE
ABOUT SYMMETRY
Ground state of a nickel(II) ion in a perfect octahedral surrounding :
3
A2 g
From the symmetry point ov view the wave - function of the ground state
can be represented as a product :
(orbital part,
part A2 g ) (spin part,
part S 1
1)).
From the tables of characters one can see that three functions corresponding
to S 1 ((or L 1,, or J 1)) form a basis for the irrep
p T1g of a cubic p
point g
group.
p
So the full ground state in spin and orbital spaces
of a nickel(II) ion in a perfect octahedral surrounding can be found
as a direct product of two irreps :
(orbital part, A2 g ) (spin part, S 1) A2 g T1g
F
From
th table
the
t bl off characters
h
t one can fnd
f d this
thi product
d t:
A2 g T1g T2 g
This is a regular rule to find qualitatively spin - orbit components in a crystal field.
field
The existence of the only irrep shows that spin - orbit coupling does not split term 3 A2 g
EFFECT OF A LOW SYMMETRY CRYSTAL FIELDFIELDAQ
QUALITATIVE ((SYMMETRY BASED)) APPROACH
Let us suppose that the symmetry is lowered, let say Oh D3d
(trigonal component of the crystal field, distortion along C3 - axis of the octahedron ) :
Spin - orbit coupling in Oh symmetry gives the only triply degenerate level ,
i.e. the quantum state belonging to T2 g irrep accordingly
to the decomposit ion of the direct product :
A2 g T1g T2 g
In order to find the effect of the trigonal field one should decompose (reduce)
the cubic irrep T2 g when the symmetry is decreased : Oh D3d .
From the characters table one finds :
T2 g Oh A1g D3d E g D3d
A1g one - dimensiona l irrep of D3d
E g two - dimensiona l irrep of D3d
The existence of two irreps shows that the trigonal crystal field together
with
ith spin
i - orbital
bit l coupling
li splits
lit cubic
bi 3A2 g term
t
i t a singlet
into
i l t A1g and
d a doublet
d bl t E g ,
this splitting exists in the absence of magnetic field - ZERO - FIELD SPLITTING
ZERO-FIELD SPLITTING: Ni2+ ION IN A TRIGONALLY
ZERODISTORTED OCTAHEDRAL SURROUNDING
Zero-field components for the ground and
excited states
PHYSICAL MECHANISM OF THE ZERO-FIELD
SPLTTING
Spin - orbital interaction does not affect (does not split) the ground state
3
A2 of a cubic system.
The trigonal field iself can not also split the ground state crystal field dos not affect spin direcly.
When spin - orbital coupling is taken into account
(like in the procedure of th evaluation of the g - factors)
the ground state gets some admixture of the excited states and
gives rise to a small orbital contribution.
In this way the ground state can be split by the trigonal
crystall field
fi ld accordingl
di ly to the
h group - theoretica
h
i l analysis.
l i
Calculation of the splitting requires application of the perturbation theory
t the
to
th degenerate
d
t ground
d state.
t t
PHENEMENOLOGICAL APPROACHAPPROACHCONCEPT OF SPINSPIN-HAMILTONIAN
The zero - field splitting can be expressed by a phenomenological Hamiltonian
that is commonly
y used in the discussionof EPR and magnetic
g
properties
p
p
.
This approach allows to avoid direct calculation of the zero - field splitting and
to express the energy levels (including Zeeman splitting )
as the functions of some phenomenological parameters.
In the case of the axial symmetry
the phenomenological Hamiltonian can be presented as :
ˆ 2 1
ˆ
H
S
S
S
1
D
ZFS
z 3
Notations:
ˆ
1) H
ZFS zero - field splittingHamiltonian,
2) Sˆ z2 operator acting on the spin - functions of the ground state,
S spin of the ground state, S 1 for Ni2 ion
3) D zero - field splitting parameter.
ABOUT THE CONCEPT OF SPINSPIN-HAMILTONIAN
" Genuine" Hamiltonia l acts in the full space
of electronic coordinate s and spin coordinate s.
SPIN - HAMILTONIAN
acts in the spin - space of a certain
orbitally
bit ll non - degenerate
d
t electronic
l t i term
t
only
l :
1
Hˆ ZFS D Sˆ z2 S S 1
3
The parameter D incorporates spin - orbital interaction that mixes
the ground state with all excited states in the crystal field,
this parameter can be estimated (only estimated but not calculated ! ) as :
D
2
,
spin orbit coupling, crystal field splitting.
SPIN--MATRICES FOR SPIN S=1
SPIN
Basis set S 1 :
MS 1 ,
MS 0 ,
M S 1 .
Three 3 3 matrices :
0 1 0
0 i
1
1
Sˆ x
1 0 1 , Sˆ y
i
2
2
0
0 1 0
0
1 0
0
0
i , Sˆ z 0 0
0
i
0
0 0 1
E
Examples
l off the
th matrix
t i elements
l
t :
1
M S 1 Sˆ x M S 1 0 , M S 1 Sˆ x M S 0
,
2
M S 1 Sˆ z M S 1 1, M S 0 Sˆ z M S 0 0 , M S 1 Sˆ z M S 1 1,
etc...
The SM S functions are the eigen - vectors of Sˆ z and Sˆ z2 ,
so the matrix of the operator Sˆ z2 is diagonal and
so,
the diagonal matrix elements are : M S2 ( in the adopted basis).
MATRIX OF ZEROZERO-FIELD AND ZEEMAN
HAMILTONIANS–– PARALLEL FIELD
HAMILTONIANS
1
Hˆ ZFS D Sˆ z2 S S 1 , Hˆ Z || B g||Sˆ zHz
3
1 0 0
1 0 0
Matrix Sˆ 0 0 0 , Matrix Sˆ 2 0 0 0
z
z
0 0 1
Matrix of Hˆ ZFS Hˆ Z || in the basis
Hˆ ZFS Hˆ Z ||
0 0 1
MS 1
MS 0
1
0
1
1
g|| BHz D
3
0
0
2
D
3
0
0
M S 1 :
1
g|| BHz D
3
The sum of the diagonal matrix elements (trace of the matrix) is zero,
0
0
1
this is due to the constant term S S 1 specially added .
3
ENERGY LEVELS - ZERO FIELD
The seqular equations :
1
DE
3
0
0
0
2
DE
3
0
0
1
DE
3
The energy levels (quantum number M S ) : E EM S :
0
0
2
1
E0 D ,
E1 E1 D ,
3
3
Triply degenerate level is split into a singlet ( M S 0) and a doublet ( M S 1).
This is in agreement with the group - theoretica l conclusion.
MS=0
|MS|=1
S=1
S=1
D
MS=0
D>0, ground singlet
D
|MS|=1
D<0, ground doublet
ENERGY LEVELS IN PARALLEL FIELD
The seqular equations :
1
g|| BHz D E
3
0
0
0
2
DE
3
0
0
1
g|| BHz D E
3
The energy levels EM S in parallel field
0
0
M S " good" quantum number in parallel field :
E0 0 ,
E1 g|| BHz D ,
E1 g|| BHz D .
The zero - field energy of the 0 component is taken as the energy origin
2
D is added to all energies, this
3
does not affect any physical results - EPR, magnetic ).
(the constant term
ENERGY LEVELS IN PERPENDICULAR FIELD
:
Th seqular
The
l equations
ti
1
DE
3
2 g x BH x 2
2
DE
3
2 g x BH x 2
0
2 g x BH x 2
DE
2 g x BH x 2
0
2 g x BH x 2
E
2 g x BH x 2
0
2 g x BH x 2 0
1
DE
3
0
2 g x BH x 2 0
DE
The energies of three levels Ei in perpendicu lar field :
1
4 g x2 B2 H2x D 2 D
2
The zero - field energy of the M S 0 component is taken as the energy origin
E1 D ,
(the constant term
E 2 E3
2
D is added to all diagonal matrix elements, i.e. to all energies).
3
FIELD DEPENDENCE OF THE MAGNETIC SUBLEVELS
ARISING FROM S=1
S=1 - LEVEL
D>0 H||C4,4
D>0,
Linear field
dependence
D>0, H||X,
Quadratic field
dependence at
low field
Main physical conclusion:
conclusion
C4 –easy axis of magnetization
PRINCIPAL SUSCEPTIBILITIES VERSUS
TEMPERATURE PLOTS FOR S=1
S 1 MOLECULE
S=
χ vs. T
D=5 cm-1
gx= gz=2
D=-5 cm-1
gx= gz=22
χT vs. T
ZERO--FIELD SPLITTING , S=1 – EPR IN PARALLEL FIELD
ZERO
+1
+1
0
-1
+1
0
S 1, D 0
-1
+1
spherical symmetry
S 1, D 0
D
D
0
0
-1
-11
H
D
axial symmetry
H
D
Scheme of the EPR transitions and spectrum for an axially
symmetric S=1 molecule, typical values of D for transition
metal complexes: 0.1- 10 cm-1
ALLOWED AND FORBIDDEN TRANSITIONS
Applied field H is parallel to axis Z,
oscillating (radiofrequency) field generating EPR transition in plane, i.e. is perpendicu lar to the main axis
Selection rules for the EPR transitions :
M 0 M 1
M 1
M 0 M 1
Forbidden transitions :
M 1 M 1 M 2
Two kinds of spectra :
1) D
and
2) D
I both
In
b th cases the
th full
f ll spectrum
t
consists
i t off two
t lines.
li
Difference :
positions
iti
off lines
li
and
d the
th changes
h
off the
th spectrum
t
with the change of frequency
RESONANCE FIELDS
R
Resonance
conditions
diti
- generall :
E M E M
M M allowed transitions
Zeeman levels : E0 0 , E1 D g BH
Case 1) D
First resonance condition :
E1 E0 D g BHres - 0 Hres
D
g B
Increase of the line move to a LOWER field
Case 1) D
Second resonance condition :
E0 E1 0 D g BHres Hres
D
g B
Increase of the line move to a HIGHER field
Case 2) D
First resonance condition :
E1 E0 D g BHres - 0 Hres
D
g B
Increase of the line moves to a HIGHER field
Case 2) D
Second resonance condition :
E0 E1 0 D g BHres Hres
D
g B
Increase of the line move to a HIGHER field
MULTI-FREQUENCY EPR, S=1
low-frequency
EPR
+1
high-frequency
EPR
+1
D
D
0
0
1
-1
2
-1
H
1 , 2 D
field
1 2
Increase of the frequencyfrequency-lines
move in opposite directions
1 , 2 D
H
1 2
Increase of the frequencyfrequency-lines
move in the region of high field
ZERO--FIELD SPLITTING , S =3/2 – EPR
ZERO
3
S , H || main axis
2
MS
MS
3
2
2|D|
1
MS
2
Three lines,,
1
1
MS ,
2
2
1
3
1
3
MS MS , MS MS
2
2
2
2
allowed transitions : M S
3
2
1
Hˆ ZFS D Sˆ z2 S S 1
3
2 5
E M S D M S
4
MS
1
2
Cr
1
MS
2
3
MS
2
3
3
t2 g
SOME BOOKS OF DIFFERENT
COMPLEXITY ON EPR
• A. Abragam and B.Bleaney, Electron Paramagnetic
Resonance of Transition Metals,
Metals Clarendon Press
Press,
Oxford, 1970
((fundamental top
p level book , the most deep
p and most
complete description of all main concepts).
• A.Carrington, A. D. McLachlan, Introduction to Magnetic
Resonance with Application to Chemistry and Chemical
Physics, Harper&Row Pub, NY, 1967
(very well designed textbook for chemists with clear
presentation of the basic principles and analysis of the
experimental data and applications)
• J. E.Wertz, J.R.Bolton, Electron Spin Resonance,
Elementary Theory and Practical Applications,
McGraw-Hill, 1972
(an introductory textbook , simple and clear book
book, contains
practical applications and tasks).
MAIN PHYSICAL CONSEQUENCES OF THE
ZERO--FIELD SPLITTING
ZERO
PLITTING
• Anisotropy of the magnetic moments and
magnetic susceptibility
• Splitting
S litti off th
the EPR spectra
t under
d the
th influence
i fl
of the non-cubic crystal fields
• Anisotropy of the EPR spectra respectively the
direction of the applied magnetic field- angular
dependence of the g-factors, positions of the
lines and their intensities
• Broadening of the EPR line in a powder sample
with a specific
p
line-shape
p
Chapter
p V.
Exchange interaction in clusters.
Exchange
E
h
effect,
ff t the
th nature
t
off the
th
potential exchange.
p
g
Magnetic properties of binuclear
compounds dimers of Cu(II)
compounds,
Cu(II), EPR
EPR,
magnetic anisotropy.
TWO ONE
ONE--ELECTRON IONS - HAMILTONIAN
The aim of this Section :
to demonstrate the main physical idea of the
exchange
h
Hamiltonian
H ilt i
( W.Heisenberg, 1926; P.A.M.Dirac, 1929)
System under consideration –
two interacting hydrogen-like atoms or ions containing one
electron each over closed ((filled)) shells.
Hamiltonian:
Hˆ Hˆ 0 Vˆ ,
Hˆ 0 Hamiltonia n of non - interactin g ions (main part)
sum off two
t intra
i t - atomic
t i interactio
i t
ti ns
Vˆ interactio n (perturbat ion)
inter - atomic interactio ns
Electrons- ”1” and “2”, nuclei- A and B (charge of the nuclei-Ze,
rA1 –position of the electron “1”
1 relatively nucleus “A”,
A , etc,
r12 - radius-vector of the electron “2” relatively electron “1”
r12
1
2
rA1
rB2
RAB
A
B
H ilt i off ttwo non
Hamiltonian
non--interacting
i t
ti
one--electron
one
l t
atoms
t
2
2
2
Ze
Ze
2
2
Hˆ 0
1 2
2m
rA1 rB 2
2 2
1 operator
t off the
th kinetic
ki ti energy off the
th electron
l t
"1" , etc
t
2m
Ze 2
operator of the potential energy of the electron "1"
1 , etc
rA1
Interatomic interaction
1
rA2
A
2
r12
rB1
rAB
B
2
2
2
2 2
e
Ze
Ze
Z
e
Vˆ
r12 rB1 rA2
rAB
e2
interelectronic ((Coulombic ) interaction ((repusion)
p
)
r12
Ze 2
attraction of electron "1" to the nucleus " B"
rB1
Z 2e 2
repulsion of the nuclei
rAB
Wave--functions of a bi
Wave
bi--atomic system
Wave - functions of the ions : A r1 and B r2 ,
these one - electron orbitals are supposed to be non - degener ate
(like 1s - orbital of a hydrogen atom ).
) They are eigen - func tions of two
Hamiltonians for two identical non - interacting ions and obey two
identical Schrodingerr' s equations
E A and EB
2 2 Ze 2
2m r A r1 E A A r1
1A
2 2 Ze 2
2m r B r2 EB B r2
2B
are the energies of the non - interacting identical ions
and , of course, they are equal : E A EB E0 .
Note :
electron "1" is " attached" to the nucleus " A" and electron "2" - to " B"
The wave - function of the Hamiltonian Hˆ 0 product :
A r1 B r2 ,
electron "1" at the nucleus " A", electron "2" at the nucleus " B".
The probability to find the electron "1"
1 in the vicinity of the point r1
(near " A" ) and the electron "2" in the vicinity of the point r2
(near " B" ) is this product raised to the second power :
| A r1 B r2 |2 | A r1 |2 | B r2 |2
In fact,
fact in the non - interacting atoms the electrons move
mo e
independently
A r1 B r2 eigen vector of Hˆ 0 :
Hˆ 0 A r1 B r2 E A EB A r1 B r2
The full energy (eigen-value) sum of the energies :
E A EB 2 E0
Notation : A r1 A 1, B r2 B 2, etc.
Let us define the function with the transposed electrons,
electrons
this function can be obtained with the aid of
transposit ion operat or Pˆ interchang e of the electrons :
12
Pˆ12 A 1 B 2 A 2 B 1
Due to indistingu ishibility of the electrons the energy
of the system with the transposed electrons
A 2 B 1
will have the same zeroth order energy 2 E0 .
The parturbati on will be :
2
2
2
2 2
e
Ze
Ze
Z
e
ˆ
V
r12 rB 2
rA1
rAB
Let us construct symmetric and antisymmet ric combinatio ns
of the bi - electronic functions A 1 B 2 and A 2 B 1
(with respect to permutatio n of the electrons) :
A 1 B 2 A 2 B 1 symmetric
A 1 B 2 A 2 B 1 antisymmet ric
They are eigen - functions of the permutation operator Pˆ12
with the eigen values : 1 and -1 respectively.
These two new wave - functions are NOT normalized .
First, let us note that the orbitals A and B localized at
different centers are non - orthogonal
g
, the overlap
p integral
g
will be denoted as AB :
AB A r B r d
Illustration for the NonNon-orthogonality of the Atomic
Orbitals
A
B
E
Electron
ic densities
Spatial distribution of the electronic densities in the orbitals
A r
B r
2
RA
A , B
2
Region of
overlap
RB
R
Normalized orbital wave-functions
with due account of the overlap
p
1
A 1 B 2 A 2 B 1 symmetric
1,2
2
2 1 AB
1,2
1
2
2 1 AB
A 1 B 2 A 2 B 1 antisym.
Exercise : find normalization factors, i.e. prove this equation
W
Wave
functions
f
ti
include
i l d only
l electronic
l t i coordinates.
di t
N
Now we
should remember about the electronic spin.
Four bielectronic spin-functions:
spin functions:
1 2 spin 1 - " up", spin 2 - " up"
M 1, S 1
1 2 spin 1 - " up", spin 2 - " down" M 0, S 0 or 1
1 2 spin 1 - " down", spin 2 - " up" M 0, S 0 or 1
1 2 spin 1 - " down", spin 2 -" down" M 1, S 1
Let us pass to the symmetric and antisymmetric
spin-functions of the whole system:
1,1 1 2
1
1 2 1 2 S 1, spin - triplet states
1,0
2
1,1 1 2
1
1 2 1 2 S 0, spin - singlet state
0,0
2
Wave-functions in this equation are normalized
Notation for the bielectronic spin-function:
S , M : S total spin, S 0 or 1, M projection of the full spin
Exercise : prove that 1,1, 1,0, 1,0
are the eigen-function of the
operator
t Sz and
d indeed
i d db
belong
l
tto M=1, -1, 0 .
Indication: apply operator Sz to spin-functions S , M . Make certain that
, Mrepresented
the operatorSzSis
by the diagonal matrix within the basis set
S , M
Important observation:
three spin functions belonging to S=1 are symmetric,
spin-function belonging to S=0 is antisymmetric
Full wave - function product :
coordinate
di t function
f
ti
spin
i - function
f
ti
1,2 1, M , 1,2 0 ,0 4 functions
symmetric
1,2 1, M , 1,2 0 ,0 4 functions
f ti
antisymmet ric
Question: are all these states allowed, i.e. are all
these states realizable in the nature?
Pauli p
principle
p
Only those quantum states of a many-electron system are
allowed for which the full wave-functions are antisymmetric
with respect to permutation in any pair of electrons.
Forbidden states, they do not exist in the nature and
we must forget about these states:
1,2 1, M , 1,2 0 ,0 4 functions
y
symmetric
Allowed (realizable in the nature) states:
1,2 1, M , 1,2 0,0 4 functions
f
i
antisymmet ric
We will deal with these states only
RESULT- INTERRELATION BETWEEN
RESULTSYMMETRY OF THE ORBITAL FUNCTION
AND FULL SPIN OF THE SYSTEM
1,2 1, M , 1,2 0,0 4 functions
antisymmet ric
antisymmet ric orbital part S 1
symmetric orbital part S 0
This is a consequense of the Pauli principle.
Energy
gy of the states becomes dependent
p
of the
full spin of the system.
CALCULATION OF THE ENERGY
Energy= diagonal matrix element of the full Hamiltonian
E S 1 1,2 1, M Hˆ 1,2 1, M
E S 0 1,2 0 ,0 Hˆ 1,2 0 ,0
The Dirac’s notations are used for the matrix elements.
The Hamiltonian is independent of spin
spin-variables
variables, so:
E S 1 1,2 1, M Hˆ 1,2 1, M
1,2 Hˆ 1,2 1, M 1, M
1,2 Hˆ 1,2
We have taken into account the normalization condition :
1, M 1, M 1
In the same way for spin - singlet state one finds :
E S 0 1,2 Hˆ 1,2
For the sake of the simplicity one can neglect the overlap :
AB 0
In this approximat
pp
ion one can find the following
g expression
p
s for
the energies of spin - triplet and spin - singlet states :
1,2 Hˆ 1,2 2 E K J S 0
0
1,2 Hˆ 1,2 2 E0 K J
S 1
Notations :
K Coulomb integral,
g
J exchange
g integral.
g
The main conclusion :
the energy does depend on the full spin of the molecule.
COULOMB INTEGRAL
2
2
K A 1 B 2Vˆ A 1 B 2 d 1d 2 A 1 Vˆ B 2 d 1d 2
Alternative notation (Dirac) :
K A 1 B 2 Vˆ A 1 B 2 , or, very short : A B Vˆ A B
2
e A 1
spatial distributi ons for the electronic densities
2
e B 2
2
2
2
2 2
electrosta tic Coulomb intercente r interactio n
e
Ze
Ze
Z
e
Vˆ
at instant positions of the electrons "1"
1 and "2"
2
r12 rB1 rA2
rAB
2
ˆ
Coulomb integral K A 1 V B 2 d 1d 2
mean (averaged over all coordinate s) value
2
off intercente
i
r Coulomb
C l b interactio
i
i n
EXCHANGE INTEGRAL
J A 1 B 2Vˆ A 2 B 1 d1d 2
Dirac's notation :
J A 1 B 2 Vˆ B 1 A 2 , or, very short : A B Vˆ BA
2
Physical
h i l sense:
A 1 B 2 Vˆ B 1 A 2
Initial ► B 1 A 2
Final ► A 1 B 2
1
►Exchange
E h
of the electrons
SPIN
S
N DE
DEPENDENCE
ENDEN E
The energy of non - interactin g atoms : 2 E0
The energy of interactio n :
E S 0 K J
E S 1 K J
K spin independen
d
d t contributi
t ib ti on,
J and J spin dependent contributi ons
The energy gap depends
on the EXCHANGE INTEGRAL only :
E S 0 E S 1 2 J
COULOMB REPULSION AND EXCHANGE
e A 1
A
2
Coulomb repulsionspin independent
e B 2
exchange
h
B
Quantum effect,
spin dependent
2
SIGN OF THE EXCHANGE INTEGRALINTEGRALA SIMPLEST CONSIDERATION
U(R)
u
0
J
3
+
R
1
g
Terms off H2-molecule,
T
l
l
spin-singlet gives a deep
minimum
RAB
Bethe s dependence (1933) of the
Bethe’s
exchange integral upon the distance
between the magnetic
g
centers:
short distance –negative,
negative
long distancedistance-positive .
I
Important:
t t role
l off bridging
b id i liligands→
d
mediate exchange
SPIN--HAMILTONIAN OF THE EXCHANGE INTERACTION
SPIN
Two one - electron ions, full spin S 0 and S 1 :
E S 0 2 E0 K J
E S 1 2 E0 K J
Physical significan ce has the energy gap
( rather than the values of the enegies separately ) :
The energy gap depends on the EXCHANGE INTEGRAL only :
E S 0 E S 1 2 J
Let us introduce the " effective" Hamiltonia n acting in spin - space :
Hˆ 2 J Sˆ A Sˆ B
The following notations are used :
Sˆ A spin operator for the ion A, Sˆ B spin operator for the ion B ,
J exchange integral, Sˆ Sˆ scalar product,
A
B
Hˆ spin - Hamiltonia n of the exchange interactio n or exchange Hamiltonia n
EXCHANGE HAMILTONIANHAMILTONIAN-EXPRESSION IN TERMS
OF THE FULL SPIN OPERATOR
Exchange Hamiltonian :
Hˆ 2 J Sˆ A Sˆ B
Let us define the full spin operator (as usually) :
Sˆ Sˆ Sˆ and Sˆ 2 Sˆ 2 Sˆ 2 2 Sˆ Sˆ .
A
B
A
B
A
B
d as :
Scalar p
product Sˆ A Sˆ B can be represente
p
ˆS Sˆ 1 Sˆ 2 Sˆ 2 Sˆ 2
A B
A
B
2
so that the exchange Hamiltonian becomes :
Hˆ J Sˆ 2 Sˆ 2 Sˆ 2 .
A
B
Important : the exchange Hamiltonian is expressed in terms of the
operators of the full spin of the system Sˆ 2
and ionic spin - operators Sˆ 2 and Sˆ 2
A
B
EIGEN--VECTORS OF THE EXCHANGE HAMILTONIAN
EIGEN
T
Two
spin
i angular
l momenta
t S A and
d S B , eigen
i
- vectors
t
are to
t be
b presented
t d
using angular momenta coupling (addition ) scheme in quantum mechanics :
S A SB S M
ith S S A S B , S A S B 1,..., S A S B
with
M quantum number of the full spin projection .
S S S M - are eigen
i
- vectors
t
off Sˆ 2 , Sˆ :
A
B
Z
Sˆ 2 S A S B S M S S 1 S A S B S M ,
Sˆ Z S A S B S M M S S A S B S M .
S A S B S M - are also eigen - vectors of Sˆ A2 , Sˆ B2 ( but NOT!!! Sˆ AZ and Sˆ BZ ) :
Sˆ A2 S A S B S M S A S A 1 S A S B S M ,
Sˆ B2 S A S B S M S B S B 1 S A S B S M .
This means that the lengths of S A and S B remain constant
but they have no definite projection s
( just like vectors L and S in the case of spin - orbit coupling).
SPIN-COUPLING SCHEME –
SPINA CLASSICAL PICTURE
S
SA
SA
Vectors SA and SB (ionic spins) precess at the conical
surfaces around the full spin vector S, only projections
SA and
d SB onto
t the
th direction
di ti off th
the vector
t S are non-zero
EIGEN--VALUES OF THE EXCHANGE HAMILTONIAN
EIGEN
Th energy levels
The
l
l can be
b found
f
d as the
th mean values
l
off the
th Hamiltonia
H ilt i n :
E S S S S M Hˆ S S S M
A
B
A
B
S A S B S M J Sˆ 2 Sˆ A2 Sˆ B2 S A S B S M
Using
g the p
properties
p
operators
p
Sˆ 2 , Sˆ A2 , Sˆ B2 one finallyy finds :
E S J S S 1 S A S A 1 S B S B 1
The energy levels are enumerated
by the quantum number of the full spin S ,
they DO NOTdepend on the spin projection M .
1
S 0 and 1.
2
3
1
E 0 J , E 1 J
2
2
The gap between two levels : E 0 E 1 2 J
S A SB
EXCHANGE SPLITTINGSPLITTING-ENERGY PATTERN
3
E 0 J
2
S=0
S=1
2J
2J
S=1
1
E 1 J
2
1
E 1 J
2
J>0- ferromagnetic
coupling,
p g high-spin
g p
ground state
S=0
3
E 0 J
2
J<0, antiferromagnetic
coupling,
p g low-spin
p
ground state
Important
p
p
preliminary
y note:
exchange interaction is magnetically isotropic
MOLECULAR STRUCTURE
OF A DINUCLEAR Cu(II) ACETATE
bridging ligands
Two Cu(II) ions are connected by the bridging ligands in a
dimer Cu-Cu
dimer,
Cu Cu separation is about 2
2.64A
64A, point symmetry D4h
Cu2+-one hole, so SA=SB=1/2, the exchange interaction in
Cu-Cu compounds proves to be always
antiferromagnetic (J<0)
Cu--Cu EXCHANGE INTERACTION
Cu
ZA,B
d A2
Cu
x y2
XA
YA
XB
Cu
YB
d B2
Scheme of the δ-overlap
of two x2-y2 –orbitals in a
dimeric cooper(II)
cluster.
l t x2-y2 –orbitals
bit l
contain strong
admixture of pp orbitals
of the bridging oxygen
atoms.
x y2
Exchange integral involving d A2
x y2
J Cu Cu d A2
x
and d B2
x y2
of Cu(II) ions :
B
B
ˆ dA
1
d
2
H
2
d
1 d 1 d 2
2
2
2
2
2
2
2
y
x y
x y
x y
TWO EXAMPLES OF DIMERIC Cu(II)
CLUSTERS WITH DIFFERENT BRIDGES
A tif
Antiferromagnetic
ti
MAGNETIC SUSCEPTIBILTY AND MOMENTS FOR A
DIMERIC CLUSTER
Molar magnetic susceptibility should be calculated for each spin - level
separately, S , and then should be averaged taking into account
the equilibrium thermal population s of spin levels ( Boltzman factor) :
Ng 2 B2
N
T
3kT
E S
exp
S
S
1
2
S
1
p
kT
S
E S
2
1
S
exp
kT
S
The energies of a dimeric cluster system are found to be :
E S J S S 1 S A S A 1 S B S B 1
After substituition of these energies into T one obtains :
J S S 1
S
S
1
2
S
1
exp
kT
2 2
Ng B S
T
3kT
J S S 1
2
1
exp
S
p
kT
S
This expression is valid for all dimeric systems, S A and S B are so far arbitrary.
EXPRESSIONS FOR A DIMERIC Cu(II) CLUSTER
1
S A S B , S 0 , 1.
2
The energies E S are S 0 , 1 :
E 0 0 , E 1 2 J , J 0 antiferromagnetic
Ng 2 B2
T
3kT
2J
6 exp
kT
2 Ng 2 B2
kT
2J
1 3 exp
kT
2J
3 exp kT
1
Magnetic
g
moment p
per a dimeric cluster
2
Neff
3kT
T g
2
2
B2
6
2J
3 exp
kT
Temperatur e dependent due to the temperatur e
dependence of the population s for the different spin - levels
WHAT ONE CAN EXPECT ?
Magnetic moment is temperature dependent !
S 0 0,
S 1 g 2 B2 1 2 2 g 2 B2
S=1
2J
S 0
S=0
Antiferromagnetic
interaction
T→0, only the ground state is
populated, magnetic moment
vanish in the case of the
antiferromegnetic exchange.
T , both
T→
b th llevels
l are equally
ll
populated, magnetic moment
takes an intermediate value
value.
LIMITING CASES
CASES--PHYSICAL SENCE
Low temperatur e :
T 0 2 0 0 only the ground level with S 0 is populated,
High temperatur e :
Both S 0 and S 1 are equally populated
3
T 2 g 2 B2
2
Thi is
This
i just
j t the
th value
l off 2 for
f two
t
non - interactin
i t
ti g
1
Cu(II) ions S A S A ,
2
in fact for an isolated Cu(II) ion one obtains :
1
3
2
4
High
g - temperatur
p
e limit is jjust doubled value, this means that equal
q
population of the levels kills the exchange effect.
2 S g 2 B2 S S 1 g 2 B2
MAGNETIC SUSCEPTIBILITY AND
MAGNETIC MOMENTS
OF A DIMERIC Cu(II) COMPLEX
Magnetic
g
susceptibility
Magnetic
moment
LEVELS--FUNCTIONS OF THE EXCHANGE INTEGRAL
LEVELS
E S
E 0
3
J
2
1
E 1 J
2
J<0
J>0
J
0
Antiferromagnetic
substances, J<0
Ferromagnetic
substances, J>0
ABOUT THE EXCHANGE SPINSPINHAMILTONIAN
Full Hamiltonian for a pair of ions acts in the full
space-spin space and coordinate space.
Exchange Hamiltonian acts in spin-space only and
gives the same energy pattern if we assume that J is
just the exchange integral . This was demonstrated by
consideration of the spin-energy levels.
Important note: spin-Hamiltonian of the exchange is
valid NOT ONLY for two one-electron ions ( when the
calculation is simple) but in much more general cases.
Chapter VI
VI.
Heisenberg-Dirac-Van
g
Vleck model
of the exchange interaction.
Conceptt off spin-Hamiltonian.
C
i H ilt i
M
Manyelectron p
problem of the exchange.
g Spinp
coupling scheme for the polynuclear
compounds Kambe’s
compounds,
Kambe s approach.
approach Trimeric
and tetrameric clusters: basic chromium
and iron acetates. EPR spectra of
polynuclear compounds
compounds.
GENERALIZATION OF THE EXCHANGE
HAMILTONIAN
We have so far considered exchange effect
for two one-electron
one electron ions
ions.
Two important generalization of the exchange Hamiltonian
((Heisenberg-Dirac-Van
g
Vleck):
)
Generalization I
The exchange spin
spin-Hamiltonian
Hamiltonian is valid for many
many--electron
ions , i.e. full spins of the ions SA and SB are arbitrary (but not
only ½. In this case the Hamiltonian retains the general form :
Sˆ A
Hˆ 2 J Sˆ A Sˆ B
and
d Sˆ are the
th operators
t
B
of the full spins of multi - electron ions and the energies are :
E S J S S 1 S A S A 1 S B S B 1
This Hamiltonian is applicable not only in the case of two
identical ions, SA=SB (homonuclear dimers) but also when the
interacting ions are different, SASB (heteronuclear
heteronuclear dimers
dimers).
Homonuclear dimer
Me
Me
Heteronuclear dimer
Me1
Me2
BINUCLERAR Fe(III) HIGHHIGH-SPIN DIMERS
(bi l i ll iimportant)
(biologically
t t)
Fe(III)( ) orbital p
picture
(high spin ion)
5
S A S B , S 0, 1, 2 , 3, 4 , 5
2
antiferrom agnetic
SCHEME OF THE EXCHANGE AND ZEEMAN
SPLITTING FOR A DIMERIC Fe(II) CLUSTER
5
S A SB ,
2
full spin :
S 0 , 1, 2 , 3, 4 , 5.
Antiferrom agnetic
exchange coupling
HETERO--BINUCLEAR MULTIELECTRONIC
HETERO
SYSTEMS
Cu 2 Ni 2
1
S A , SB 1
2
Cu 2 Fe3
1
5
S A , SB
2
2
Fe3 Ni 2
5
S A , SB 1
2
Generalization II
The exchange
g spin-Hamiltonian
p
is applicable
pp
not only
y for
the dimeric systems but also for the polynuclear clusters.
clusters
The general form contains summation over all pairs of the
ions in a polynuclear system:
Hˆ 2 J ij Sˆ i Sˆ j ,
i, j
i , j numbers of the ions.
Three - nuclerar
n clerar system,
s stem general case,
case J12 J13 J 23 :
Hˆ 2 J Sˆ Sˆ J Sˆ Sˆ J Sˆ Sˆ
12 1 2
13 1 3
23 2 3
2
J23
J12
1
J13
3
Homo- and heteronuclear systems.
HOMO- AND HETERONUCLEAR
HOMOTRIMERS-- HAMILTONIANS
TRIMERS
2
J
J
J
1
3
Hˆ 2 J Sˆ 1Sˆ 2 Sˆ 1 Sˆ 3 Sˆ 2 Sˆ 3
2
2
J1
1
J2
3
J2
Hˆ 2 J1 Sˆ 1 Sˆ 2 J 2 Sˆ 1 Sˆ 3 Sˆ 2 Sˆ 3
1
J12
J23
J13
3
Hˆ 2 J12 Sˆ 1 Sˆ 2 J13 Sˆ 1 Sˆ 3 J 23 Sˆ 2 Sˆ 3
HEISENBERG - DIRACDIRAC- VAN VlECK
HAMILTONIAN
Hˆ 2 J ij Sˆ i Sˆ j
i, j
Sˆ i full spin operators,
J ij netork of the many - electron exchange parameters
Heisenberg - Dirac
Dirac-- Van Vleck model
model,
HDVV--model
HDVV
Important: this model describes molecules
(finite number of magnetic centers)
and magnetic solids
HIGH--SPIN DIMERS
HIGH
Sˆ A
Hˆ 2 J Sˆ A Sˆ B
and Sˆ are the operators
B
of the full spins of multi - electron ions and the energies are :
E S J S S 1 S A S A 1 S B S B 1
1
Ni
S A SB 1
2
Ni 2 dim er ,
S 0 , 1, 2
2
S A SB 3 2
Cr
3
Cr 3 dim er ,
S 0 , 1, 2 , 3
Intervals between the exchange levels for the dimeric clusters :
E S E S 1 J S S 1 S 1S 2 JS
E S E S 1 2 JS
Lande' s rule
TWO EXAMPLESEXAMPLES- EXCHAGE LEVELS OF
ANTIFERROMAGNETIC DIMERS
S 3
6J
S 2
S 2
4J
S 1
4J
S 1
2J
S 0
2J
S 0
2
2
Ni Ni
S A SB 1
Cr 3 Cr 3
S A SB 3 2
EPR OF A COOPER(II) DIMERDIMER-ANTIFERROMAGNETIC
EXCHANGE
+1
S=1
D
2|J|
0
-1
EPR-transitions,
EPR
t
iti
low frequency
case
S=0
S
0
Exchange Exchange Exchange+
zero field zero
zero-field+
field
+zero-field
Zeeman
Main feature: EPR from
the excited level
possessing S=1. Ground
sate with S=0 (nonmagnetic)
ti ) is
i EPR silent
il t ,
this is the main
q
of the
consequence
antiferromagnetic
exchange. Intensities of
the lines are
temperature dependent
–proportional to the
population of the excited
state, i.e. increase with
the increase of the
temperature.
TRIMERIC CLUSTERS: HOMOHOMO- AND
HETERO--NUCLEAR TRIMERIC CARBOXILATES
HETERO
S Cr
3
S Fe3
3
2
5
2
-metal
-O, C
Triads of the metal ions :
Cr 3 Cr 3 Cr 3 Cr3 , Fe3 Fe3 Fe3 Fe3 ,
Fe3 Cr 3 Cr 3 FeCr2 , Fe3 Fe3 Cr 3 Fe2Cr
POLYOXOANION [NiNa(H2O)2(AsW9O34]11WO6
NiO6
Na
AsO4
Trimeric
3Ni2+magnetic
f
fragment
m t
S Ni 2 1
S1 S 2 S3 1
Polyhedral representation
SPIN-COUPLING SCHEME
SPINFOR A TRIMERIC SYSTEM
Three spins : S1 , S 2 and S3
Successive spin coupling according to the general rule :
Stage 1. Coupling of two spins S1 and S 2
to get spin S12 int ermediate spin :
S12 S1 S 2 , S1 S 2 1, , S1 S 2
Stage 2. Coupling of two spins S12 and S3
to get spin S123 S full spin :
S S12 S3 , S12 S3 1, , S12 S3
QUANTUM NUMBERS IN THREE
THREE--SPIN
COUPLING SCHEME
Th
Three
quantum
t
numbers
b
i the
in
th addition
dditi scheme
h
for three arbitrary spins :
S12 intermedia te spin,
S full spin,
spin
M full spin projection
:
Di ' s notations
Dirac'
t ti
(l b l ) for
(labels)
f the
th eigen
i
- vectors
t
S12 S M
2 ˆ2
Eigen functions of Sˆ12
, S and Sˆ z
EIGEN--VECTORS, THE MAIN PROPERTIES
EIGEN
S12 S M
2
eigen - vectors of three operators Sˆ 2 , Sˆ12
and Sˆ z :
Sˆ 2 S12 S M S S 1 S12 S M ,
2
S12 S M S12 S12 1 S12 S M ,
Sˆ12
Sˆ z S12 S M M S12 S M
These equations mean that full spin ,
intermedia te spin and
full spin
p p
projection
j
have definite values
in S12 S M state of a three spin system
NOTE : S12 S M is
i not an eigen
i
- vector
t off Sˆ12 z operator
t
SPIN LEVELS FOR A SYMMETRIC
HOMONUCLEAR
H
M N LE
TRIMER
ME
2
J
J
1
3
J
HDVV Hamiltonia
a to an for
o a sy
symmetric
et c ttrimeric
e c ccluster
uste :
Hˆ 2 J Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ
1 2
1 3
2 3
Full spin operator :
Sˆ Sˆ Sˆ Sˆ
1
2
3
2
Sˆ 2 Sˆ 1 Sˆ 2 Sˆ 3 Sˆ12 Sˆ22 Sˆ32 2 Sˆ 1Sˆ 2 Sˆ 1Sˆ 3 Sˆ 2 Sˆ 3
2 Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ 2 Sˆ 2 Sˆ 2 Sˆ 2
1 2
1 3
2 3
1
2
3
Expression for the exchange Hamiltonian in terms of the
full spin
p operator
p
:
Hˆ J Sˆ 2 Sˆ 2 Sˆ 2 Sˆ 2
1
2
3
ENERGY LEVELS OF SYMMETRIC TRIMERS
Symmetric homonuclea r trimer J12 J13 J 23 :
Hˆ J Sˆ 2 Sˆ 2 Sˆ 2 Sˆ 2
S12 SM
1
2
3
are the eigen - vectors of the Hamiltonia n.
C l l ti n off the
Calculatio
th energy levels
l
l :
S12 SM Hˆ S12 SM S12 SM J Sˆ 2 Sˆ12 Sˆ 22 Sˆ32
S
J S S 1 S1 S1 1 S 2 S 2 1 S 3 S 3 1
12
SM
The energy levels for a symmetric trimer :
E S J S S 1 S1 S1 1 S 2 S 2 1 S 3 S 3 1
Note : the
th energies
i do
d DEPEND the
th FULL SPIN
S
l
only,
they are INDEPENDEN T of the INTERMEDI ATE SPIN ,
this result is applicable for the symmetric trimers only.
EXAMPLE: A TRICOOPER CLUSTER (Si=1/2)
1
Tri - cooper II clusters, S1 S 2 S3 .
2
Stage 1. S12 0 and 1
Stage
g 2.
1
1
3
S12 0 S ; S12 1 S
and S
2
2
2
Full set of quantum numbers
defining
d
fi i full
f ll spin
i off a trimer
ti
:
S12 S int ermediate spin full spin
For a tri - cooper(II) cluster :
S12 S 0 1 , 1 1 , 1 3
2
2
2
1
Important
po a : two
os
states
a es with S a
and
dd
different
e e intermedia
e ed a te
e sp
spins
s
2
SYMMETRIC TRI-COOPER(II) CLUSTER,EPR
B Cage,
B.
C
F.
FA
A. C
Cotton,
tt N S
S. D
Dalal
l l ett al,
l J.Am.Chem.Soc.,2003
J.Am.Chem.Soc.,
J A Ch S 2003
3
S
2
3||J||
Zero-field splitting
Exchange splitting ( -3J = 321K)
1
S
2
(a) The molecular structure of Cu3(O2C16H23)6.
The arrows indicate the equilateral triangle formed by three Cu2+ ions separated
by 3.131 and bridged by two carboxylate groups.
(b) Energy level diagram and the expected EPR transitions
in the HDVV scheme for a symmetric tri-cooper cluster.
MAIN FEATURES OF THE EPR OF THE
SYMMETRIC TRIMERS IN THE HDVV MODEL
• A single
g line arising
g from the intra
intra--doublet transition in
the ground state, S=1
S=1/2 (line 2’)
• Three lines (1, 2 and 3) from the excided level that is
split:: combined effect of the zero
split
zero--field splitting and
Zeeman splitting ( full spectrum consists of three lines
lines,,
lines 2 and 2’ have the same resonance fields)
• Intensity of the line 2’ decreases with the increase of
the temperature , lines 1, 2 and 3 increase the
intensities with the increase of the temperature
(proportional to Boltzman population of the level that is
relevant to a specific line in EPR)
• EPR spectrum is strongly anisotropic , i.e. depends on
the orientation of the magnetic field respectively
molecular axes
axes..
TRIMERIC COOPER(II) CLUSTERCLUSTERA NEW EXAMPLE
Spin arrangement
Excited state
Inorg.Chem. 41 (2002) p.5821
Ground state
Problem of “spin frustration” -degenerate ground state
EXAMPLE: TRICHROMIUM CARBOXILATE (Si=3/2)
3
Tri - chromium III clusters
clusters, S1 S 2 S3 .
2
Stage 1. S12 0, 1, 2, 3.
3
1 3 5
Stage 2. S12 0 S ; S12 1 S , , ;
2
2 2 2
1 3 5 7
3 5 7 9
S12 2 S , , , ; S12 3 S , , , .
2 2 2 2
2 2 2 2
F ll sett S12 S int
Full
i ermediate
di spin
i full
f ll spin
i :
For a tri - chromium(I II) cluster :
S12 S
1
1
3
3 3
3
5
5
5
7
7
9
1 , 2 ; 0 ,1 2 , 3 ; 1 ,2 , 3 ; 2 ,3 ; 3 .
2
2
2 22
2
2 2
2 2
2
2
S
1
2
S
3
2
Important : two states with S
S
5
2
S
7
2
1
3
, four states with S ,etc
2
2
S
9
2
ENERGY PATTERN OF A SYMMETRIC TRIMERIC
Cr(III) CLUSTER
E S J S S 1 S1 S1 1 S 2 S 2 1 S3 S3 1
45
J S S 1
4
L d ’ rule:
Lande’s
l
E S E S 1
2 JS
_____________
Degeneracy with
respect to the
intermediate
spin value
S
9
2
3 9
2
9J
S
2 7 ,3 7
7
2
2
2
7J
S
5
2
3
2
1
S
2
1 5 ,2 5 , 3 5
5J
S
3J
2
2
2
0 3 ,1 3 ,2 3 , 3 3
2
2
1 1 , 2 1
2
2
2
2
SPIN LEVELS
FOR
FO A HE
HETERONUCLEAR
E ON LE
TRIMER
ME Me2 Me
Me’’
2
J1
1
J2
3
J2
HDVV Hamiltonia n for a heteronucl ear trimeric cluster :
Hˆ 2 J Sˆ Sˆ 2 J Sˆ Sˆ Sˆ Sˆ
1 1 2
2
1 3
2 3
:
Equivalent
E
i l t form
f
Hˆ 2 J 2 Sˆ 1 Sˆ 2 Sˆ 1 Sˆ 3 Sˆ 2 Sˆ 3 2 J1 J 2 Sˆ 1 Sˆ 2
Full spin operator :
Sˆ Sˆ Sˆ Sˆ
1
2
3
ˆS 2 Sˆ Sˆ Sˆ 2 Sˆ 2 Sˆ 2 Sˆ 2 2 Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ
1
2
3
1
2
3
1 2
1 3
2 3
2 Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ 2 Sˆ 2 Sˆ 2 Sˆ 2
1 2
1 3
2 3
1
2
3
Intermediate spin operator :
Sˆ Sˆ Sˆ
12
1
2
ˆS 2 Sˆ Sˆ 2 Sˆ 2 Sˆ 2 2 Sˆ Sˆ
12
1
2
1
2
1 2
2 Sˆ Sˆ Sˆ 2 Sˆ 2 Sˆ 2
1 2
12
1
2
Full Hamiltonian in terms of the full spin
and intermediate spin operators :
Hˆ 2 J 2 Sˆ 1 Sˆ 2 Sˆ 1 Sˆ 3 Sˆ 2 Sˆ 3 2 J1 J 2 Sˆ 1 Sˆ 2
2
J 2 Sˆ 2 Sˆ 12 Sˆ 22 Sˆ 32 J1 J 2 Sˆ 12
Sˆ 12 Sˆ 22
:
Eigen values
Ei
l
E S12 , S J 2 S S 1 S1 S1 1 S 2 S 2 1 S3 S3 1
J J Sˆ Sˆ 1 S S 1 S S 1
1
2
12
12
1
1
2
2
E S12 , S depend on full and intermedia te spin quantum numbers.
Kambe' s approach.
EPR OF TRIMERS WITH HALF
HALF--INTEGER SPINS–
SPINS–
GROUND TERM (HDVV MODEL)
symmetric
J1
J
J
distorted
J1
J
J2
J
S12 1
S12 1 , S12 1
2
2
2
H
1
S12
2
H
The only line within the HDVV model. Anisotropic exchange
interactions result in a much more complicated spectrum
spectrum,
see a comprehensive review paper (next page).
REVIEW OF THE
ANISOTROPIC
EXCHANGE
INTERACTIONS
AND EPR
BIOLOGICAL
BIOLOGI
L SYS
SYSTEMS
SYSTEMSEMS-TWO
WO EX
EXAMPLES
M LES
S-cys
S
Fe
Schematic structure of the
protein with [Fe3S4] core.
core
S-cys stands for the sulfur atom
of a cystein group.
Th
Three
magnetically
ti ll coupled
l dF
Fe iions.
Schematic structure of the
two iron (Fe2+, Fe3+ ) ferredoxin
two-iron
ferredoxin.
S-cys stands for the sulfur atom
of a cystein group.
T
Two
magnetically
ti ll coupled
l dF
Fe iions.
TETRANUCLEAR METAL CLUSTERS
Teranuclear cluster MnCu3 with a central magnetic ion
ˆ 2 JSˆ Sˆ Sˆ Sˆ
H
A B1
B2
B3
MnCu oxpn 3
with oxpn oxamido N , N 3a min opropane
from the book of O.Kahn
Scheme of the exchange
pathways in the exchange
Hamiltonian
Schematized structure of the
tetranuclear cation MnCu3
CUBANE--LIKE TETRANUCLEAR SPECIES
CUBANE
Teranuclea r cluster Me4 , four metal ions occupy the
vertices A, B, C, D of a regular tetrahedro n,
exchange Hamiltonia n contains the only exchange parameter J :
Hˆ 2 J Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ Sˆ
A
B
Idealized structure:
A C
A
D
B
C
B
D
C
D
Tetranuclear Cu(II) -bridged dimers linked
through weak Cu-bridge interactions
POLYOXOANION [Ni6 As3W24O94(H2O)17]WO6
Inorg. Chem. ,2003, 42, 5143-52
NiO6
AsO4
Polyhedral representation
Two coupled
T
l d
trinuclear
3Ni2+magnetic
fragments
Cr8-MOLECULE,
MOLECULE GROUND STATE
STATE--S=0
http://cmpweb.ameslab.gov/magnetic_molecules/cr812.html
EXAMPLES OF MORE COMPLICATED
POLYNUCLEAR SYSTEMS
Three trimeric NiII clusters incorporated
into polyoxomet alate structure
EXTENDED SYSTEM ( ONEONE-DIMEMSIONAL ) MAGNETIC CHAIN
Structure of the infinite linear chain polyanion
[MnPM11O39] (M=W, Mo) exhibiting magnetic coupling
between Mn ions
J.M.Clemente-Juan, E.Coronado,
Coordination Chem.
Chem Rev.,
Rev 193-195
193 195 (1999) pp.361-394
pp 361 394
Chapter
Ch
t VII
VII.
Single molecule magnets.
magnets
Physical principles- quantum tunneling,
relaxation Mn12
relaxation.
Mn12-ac
ac molecule
molecule.
Applications in molecular electronics.
SINGLE MPLECULAR MAGNETS (SMM) EXCELLENT REVIEW OF THE FOUNDERS OF THE FIELD
• Fundamental physical
concepts needed to
understand the
phenomena of single
molecular
l
l magnetism
ti
and
d
quantum size effects
•Present day state off the
field: what has been done
and critical discussion
discussion,
correlation between
structure and p
properties
p
of
the molecules
•Perspectives
Perspectives
Hereunder in the description of SMM problem I follow mainly this paper
SHORT
PRESENTATION
OF THE
UNDERLYING
CONCEPTS
RELAXATION--CLASSICAL
RELAXATION
H
Magnetic field -give
rise to spin ordering
Relaxation time - mean time of
spin reordering after switching
off the magnetic field
Isolated spins,
p
molecular systems
y
–short time ( 10-6 sec),
)
solid state magnets
magnets-- long time ( practically- infinite).
object)- ”forgets”
forgets the direction of the
•Isolated spin (quantum object)applied field during the time about 10-6 sec
•Solid state magnet (classical object) - “memorizes”
memorizes applied
field “forever
forever”
SINGLE MOLECULAR MAGNETSMAGNETSDISCOVERY
DIS
OVERY OF THE
HE PHENOMENON
HENOMENON
[Mn12O12 (CH3COO)16 (H2O)4] -molecule - Mn12-ac (Mn12-acetate)
4 S Mn 4
ferromagne tic
Antiferromagnetic
coupling between
M 3+ and
Mn
dM
Mn4+
Mn4+
Mn 3+
8 S Mn 3
f
ferromagne
ti
tic
MANGANESE--12 CLUSTER
MANGANESE
eight Mn33+ ions (Si =2)
and four Mn4+ ions (Si =3/2)
Ground state S=10,,
magnetic moment
20B
DISCOVERY
OF SINGLE MOLECULAR MAGNETISM
Basic papers:
• A.Ganeschi,
AG
hi D
D.Gatteschi,
G tt
hi R
R.Sessoli,
S
li
A.L.Barra, L.S.Brunel, M.Guillot,
J.Am.Chem.Soc, 113 (1991) 5873.
• R.Sessoli,
R Sessoli D
D.Gatteschi,
Gatteschi M
M.A.Novak,
A Novak
Nature, 365 (1993) 141.
Mn12--ac MOLECULE
Mn12
MOLECULE--SLOW RELAXATION
Large molecule – object possessing intermediate properties,
between quantum and classical ones.
ones Mn12-ac
Mn12 ac molecule
shows slow relaxation of the magnetization at low temperature.
If the Mn12-ac molecule is magnetized
g
byy an applied
pp
field,,
the molecule retains magnetization for a long time,
approximately 108 seconds = 3 years at 1.5K . Under this
condition a single molecule becomes like a tiny magnet, in
the sense that if magnetized by an applied field it “remember”
magnetization for days or months.
months Under this respect
therefore Mn12-ac molecule behaves like a classical magnet.
Applications-molecular electronics:
• memory storage elements in one molecule
• quantum computing – memory units of molecular size
HYSTERESIS LOOPS OF Mn12Mn12-ac
AT DIFFERENT TEMPERATURES
Experiment: L.Thomas et al. Nature, 383 (1996) p.145
Magnetic hysteresis – evidence of slow relaxation,
relaxation
condition for storing information in a particle
particle.
Mn12-ac can be referred to as a single molecule magnet
potential energ
gy
QUANTUM TUNNELING
IN A DOUBLE WELL SYSTEM
a) non
non--interacting states
states, double
degenerate quantum level, each
corresponding
di tto a localized
l
li d state
t t
in the left and in the right wells
b) interacting states,
states giving rise to
tunnel splitting
p
g T:
T=hν, ν- is the frequency of
tunneling through the barrier.
Double-well potential:
Doublequantum and classical pictures
ABOUT SOME CONCEPT OF QUANTUM
MECHANICS IN A FEW WORDS
•Macroscopic ( classical objects).
The macroscopic
p objects
j
mayy be stable in two different
states, but they can have only one state at a time
time. If
we consider a ball in a container characterized by two
wells, Fig. (a), it may be either in the left or in the right
well, but once a choice is made it is clear that its state
i described
is
d
ib d , for
f instance,
i t
b the
by
th statement:
t t
t the
th ball
b ll is
i
in the right well. It can change its state, by overcoming
the barrier T separating two wells (jumping over the
barrier), and then roll down into the left well.
•Microscopic ( quantum objects).
A quantum object has also a wave nature
nature, and if the wavefunction of the left-hand particle extends over to the rightparticle must be
hand well, and vice versa, the state of the p
described by a superposition of the two states:
(
) ( ) - (right)]
( g )
u=(1/2)[(left)
g=(1/2)[(left) + (right)]
Since the wave
wave-function
function of the left well extends to the right
well with a nonzero value, the probability of observing the
left ball in the right well is different from zero. The main
result of quantum-mechanical consideration: a “quantum”
ball can be both in the right- and in the left-hand well. The
particle can pass from the left to the right and back without
climbing the barrier, but tunneling. This effect is called
quantum
qua
u tunneling,
u e g, thiss iss o
one
e o
of the
e most
os spec
spectacular
acu a
manifestations of quantum mechanics.
N
Example
E
l off quantum
t
tunnelingg
ammonia molecule,
two positions of N, inversion of
the molecule.
The ground state is splitinversion splitting
splitting,
IR spectroscopic manifestation.
H
H
H
The probability of tunneling exponentially decreases with
the increase of the barrier height and the particle mass.
Therefore it is expected to be most observable in small
particles at low temperature.
ROLE OF THE ENVIRONMENT
When th
Wh
the molecule
l
l iis separated
t d ffrom th
the environment,
i
t
only intramolecular interactions are involved the tunneling
occurs without loss of the energy
energy. Coupling to the
environment ( thermostat) means that the particle loses
energy with the tunneling.
Tunneling from a
metastable state.
In the case of strong coupling with the environment , that is
much larger than the tunnel splitting, the particle will stay
localized in one of two wells, and will not be able to tunnel.
F the
For
th intermediate
i t
di t coupling
li
th
the particle
ti l can ttunnell b
butt th
the
coherence of the motion between wells will be lost.
JAHN-TELLER DISTORTIONS IN
JAHNM
Mn12
Mn1212-ACETATE
CET TE MOLECULE
Angewandte. Chem. , 2005
Effect of Pressure on the Magnetic Anisotropy in the Single-Molecule Magnet
Mn12-acetate: An Inelastic Neutron Scattering Study
Andreas Sieber, Roland Bircher, Oliver Waldmann, Graham Carver,
Grégory Chaboussant, Hannu Mutka, Hans-Ulrich Güdel
ZERO--FIELD SPLITTING HAMILTONIAN
ZERO
FOR Mn12
Mn12--ac
Ground spin state for Mn12-ac molecule : S=10.
S=10
Zero-field splitting Hamiltonian:
H ZFS
ˆ 2 1
ˆ 2 110
D S z S S 1 H ZFS D S z
3
3
D zero - field splitting parameter.
parameter
Zero - field splitting removes the degeneracy of spin state S :
110
E M S D M S2
S MS S
,
3
For Mn12-ac molecule D is NEGATIVE, under this condition
the M S S levels will lie lowest.
ENERGY LEVELS
2 110
E M S D M S
,
3
S MS S
Set of 2 S 1 21 levels,,
10 double degenerate levels
M S 6
M S 7
M S 8
M S 9
M S 10
(energies are independen t
of the sign of M S ) ,
one levels M S 0
non degenerate .
D negative for Mn12 - ac,
ground state with M S 10
ZEEMAN SPLITTINGSPLITTING-GROUND LEVELS OF Mn12
Mn12
ˆ 2 1
H ZFS D S z S S 1 g B Sˆ zHz
3
Zero - field splitting effect and Zeeman interaction
completely removes the degeneracy of spin state S :
M S 8
110
E M S D M S2
g B M S H z
3
M S 10
M S 9
negative D ,
maximum spin projection
in the g
ground state :
M S 10
D 0,
S MS S
M S 10
ENERGY LEVELS FOR A SPIN STATE S WITH
EASY AXIS OF MAGNETIC ANISOTROPY
The MS levels are located in the left (
)well
and the –MS levels in the right( ) well
well.
(a) In zero field the levels are double
d
degenerate
t and
d equally
ll populated
l t d
(the have the same energies)
(b) The application of a magnetic
field selectively populates the
right
i h wellll (the
( h energy iin the
h fifield
ld
becomes lower)
(c) After removing the magnetic
field the molecule returns to
equilibrium
ilib i
th
through
h a series
i off
steps
MAGNETIZATION AND RELAXATION
Mn12-ac molecule is characterized by magnetic
anisotropy along “easy
easy axis
axis”, that means that the
magnetization is preferentially oriented parallel to Z-axis (a).
pp
parallel to Z-axis the levels with
p
When a field is applied
positive MS correspond to a projection of magnetization
antiparallel to field, while those with negative MS correspond
t magnetization
to
ti ti
parallel
ll l to
t the
th applied
li d external
t
l field
fi ld (b).
(b)
The separation in zero field between |MS| and |MS-1|
levels is given by (2|MS|-1)D
| 1)D . The system can be prepared
in a magnetized state by applying a magnetic field parallel to
Z-axis.
Z
axis. If temperature is low and the magnetic field is large
the MS=-10 state will be the only one populated and the
magnetization reaches the saturation value.
When the field is removed the system
y
must g
go back
to thermal equilibrium-”relaxation (c). This means that at the
equilibrium half of the molecules must be in the MS=-10 and
h lf in
half
i the
th MS=10
10 state,
t t with
ith no resulting
lti magnetization.
ti ti
Th
The
relaxation ( the return to the equilibrium) can be controlled
by measuring the magnetization as a function of time.
time For
some systems the relaxation obeys the exponential low. In
the case under consideration this means that the
magnetization decreases after the field is switched of as:
t
M z t M z t 0 exp
τ
where is the “relaxation time”-characteristics of the rate.
Mechanism of the relaxation- interaction of spin with the
environment
i
t ( phonons
h
–lattice
l tti vibrations).
ib ti
)
PHONON--ASSISTED TUNNELING RELAXATION
PHONON
Absorption of the phonons from the crystal surrounding leads to the
population of the excited magnetic levels, this decreases the
effective barrier for the reversing the magnetization and in this way
increases the rate of tunneling .
A possible “short cut” to magnetic relaxation through
tunneling between thermally activated states
states.
OTHER SINGLE MOLECULAR MAGNETS
C
O
Fe4-molecule
Fe8-molecule
Schematic structure , preferred orientations of
individual spins are pointed out
Energy of mag
gnetic a
anisotrropy
Semiclassical Picture of Spin Turning
Wh Crossing
When
C
i the
th B
Barrier
i
DS2
M=-S
M=0
M=S
M
(spin projection
projection-magnetic
magnetic moment)
Barrier U(M)
( )
for the reversal of
magnetization
g
in a
high-spin magnetic
molecule,
height of the barrier
2.
U(M=0)=DS
(
)
In the absence of
the magnetic
g
fieldfieldtwo minima:
U(M=-S)=
(
) U(M=S)
(
)
“Classical” spinspin-vector with a certain direction
Energy levels of the S=10
spin manifold split by an
axial anisotropy (top).
Overcoming of the barrier
can occur through a thermal
activation or through a
tunnel mechanism involving
the ground doublet or
thermally excited states.
When an axial field is
applied the levels on the
opposite sides of the barrier
are no more in coincidence
(b) and tunnelling is
suppressed unless specific
values of the field are
reached (c).
Europhysics News (2003) Vol. 34 No. 2
Quantum tunnelling of the magnetisation in molecular nanomagnets
R Sessoli
R.
Department of Chemistry, University of Florence and INSTM, 50019
Sesto Fiorentino, Italy
View of the structure of the Fe8 molecular cluster
The
e iron
o atoms
ato s (ye
yellow
o )
carry the magnetic moments
that in the ground state are
arranged
g to give
g
S=10. The
shadows around the cluster
represent the actual dimensions
of the atoms and give an idea of
how the central magnetic core is
surrounded by an organic
shell.
Europhysics News (2003) Vol. 34 No. 2
Quantum tunnelling of the magnetisation in molecular nanomagnets
R. Sessoli
Sesso
Department of Chemistry, University of Florence and INSTM, 50019
Sesto Fiorentino, Italy
Single
g Molecule Magnets
g
(SMM’s):
Bistable Magnetic Units
• SMMs
are magnetically
ll
bistable systems that
require an applied field to
invert their magnetization
direction below a
“bl ki ” temperature.
“blocking”
t
t
• Bistability
(SMM) stems from
l
large
spin
i [S = 10] and
d
negative magnetic anisotropy.
Barrier height E = S2 lDl
“Frozen” superparamagnetic
p p
g
states
Mn21--clusterMn21
cluster-Single Molecule Magnet
behavior,
behavior
Inorg Chem, Ceorge Chritou, 2004,v.43,pp 4137-44
Magnetization vs field at different
temperatures, hysteresis loops
Mn22 clustercluster chain-like
chain like SMM
Hysteresis loop in sweeping
magnetic
ti field
fi ld (0
(0.07
07 T/s)
T/ )
-temperature dependence
George Christou,
Christou Inorg Chem
Chem, 2004
2004,v.43,
v 43 p
p. 4203
HIGH--SPIN CHROMIUM MOLECULES
HIGH
Cr4-molecule
Cr8-molecule
HIGH--SPIN Fe MOLECULES
HIGH
Fe8-molecule
Fe10-molecule
VANADIUM-15 MOLECULE AND
VANADIUMA NEW Fe
Fe--8 WHEEL
V15-molecule
Fe8-wheel
MIGHT A MOLECULAR SPIN CLUSTER SERVE AS A
COMPUTER ELEMENT ?
Allowing for a distance 5nm between neighboring spins, a disc with
the area 100cm2 will hold:
100cm2 / (510-7 cm)2 spins =4 1014spins
The state MS=S of each cluster (spin) would be used to
store a classical bit (remembers magnetization !!!):
1 spin 1bit
4 1014spins 4 1014bits= 4 1014/8 bytes= 50000
gigabytes
The disc holds a staggering
gg
g amount of memory:
y
50 000 gigabytes !!!
However ((!!!)) q
quantum tunneling
g renders these two states unstable,,
even at absolute zero temperature. At T=1.5K the relaxation time for
Mn12-ac is 108s (3 years). This is not enough for computers
elements (even if the refrigeration problem would be solved)
solved). An
acceptable relaxation time: at least 15 years at room temperature.
Second key problem: reading and writing bits (information)
THE MAIN TRENDS IN NANOCHEMISTRYNANOCHEMISTRYDESIGH OF NEW SMM WITH HIGH
ANISOTROPY
How to reach this goal:
• T
To increase
i
the
th number
b off the
th iinteracting
t
ti
spins in order to accumulate single-ion
anisotropy
i t
in
i a llarge cluster
l t
• Increase the anisotropy
y of the individual
ions in a controllable way (orbital
g
strongly
g y anisotropic)
p )
magnetism• Symmetry of the molecule-easy axis of
magnetization
Anisotropy
Michel Verdaguer, University Pierre&Marie Curie, Paris
Search for single molecular magnets
magnets--cyanometalates family
CoCu2
CoCo2
CoNi2
CrNi
C
N2
High spin
CrNi
7/2
5/2
CoCu3
C
Cyanometalates
t l t
CrMn6 27/2
CoCo3
CoMn6
CoNi3
CrCu6 9/2
CrNi6 15/2
CoCo6
CoNi5
CoCu6
PENTANUCLEAR Mn2(III) Mn3(II) CLUSTERS EXHIBITING
SINGLE MOLECULAR MAGNET BEHAVIOR
Kim R. Dunbar , Texas A&M Universrsity
CN
CN-group
Mn(III),
( ), d4, S=1
Mn(II), d5, S=5/2
Molecular structure
Mn5-cyanometalate
y
fragment
g
Kim Dunbar et al, Angew. Chem. Int. Ed. 42 (2003) 1523-1526
THE STRUCTURE OF [Mo75Fe30] CLUSTER.
THE YELLOW CIRCLES ARE IRON(III)
( ) IONS
http://66.102.9.104/search?q=cache:1cV6p8iEycYJ:www.europhysicsnews.
com/full/24/article4/article4.html+Molecular+Magnetism&hl=en&ie=UTF-8
/f ll/24/ ti l 4/ ti l 4 ht l M l
l M
ti &hl
&i UTF 8
Dante Gatteschi
GIANT MOLIBDENIUM OXIDE CLUSTER
CONTAINING 368 (!!!) METAL IONS
Achim Müller et all, Angew. Chem.2002.
Along the C4 axis
Perpendicular to the C4 axis
Building blocks (units ):
64 {Mo1}-yellow, 32{Mo2}-red, and 40{Mo-(Mo)5}-blue
NEW TREND IN NANOCHEMISTRY
NANOCHEMISTRY:
GIANT MAGNETIC METAL CLUSTERS
N
New
T
Trend
nd of
fN
Nanochemistry
n h mist
A. Müller,
A
Müll E
E. B
Beckmann,
k
H
H. Bö
Bögge, M
M.
Schmidtmann, A. Dress
“Inorganic Chemistry Goes Protein
Size:
S
e A Mo368
o368 Nano-Hedgehog
a o edge og
Initiating Nanochemistry by Symmetry
Breaking
Breaking”
Angew. Chem. Int. Ed. 41, 1162-1167
(2002)
Angew. Chem. 114, 1210-1215 (2002)
NEW GIANT MOLECULESMn-CLUSTERSE
EXHIBITING SINGLE
MOLECULAR MAGNET
BEGHAVIOR
Wheel-Shaped [Mn12]
Single-Molecule Magnets
Evan M.
M Rumberger,
Rumberger Sonali J
J.
Shah, Christopher C. Beedle,
Lev N. Zakharov,
Arnold L. Rheingold,
g
and David
N. Hendrickson*
Inorganic Chemistry, 2005
Mn84
Mn
84 - cluster
G.Christou
Scheme of the light induced interconversion of
P
Prussian
i blue
bl derivatives
d i ti
((See
(S
See
S nextt slide)
lid )
From: Dante Gatteschi:
http://66.102.9.104/search?q=cache:1cV6p8iEycYJ:www.europhysicsnews.com/full
/24/article4/article4.html+Molecular+Magnetism&hl=en&ie=UTF-8
Important feature of molecular magnets is that they are in general isulators,
therefore they are much more transparent to UV-visible light than classic
magnets. Therefore it is possible to use light to induce magnetic
transitions ( groups of Verdaguer and Hashimoto ). Prusian blue
derivatives are complex cyanides of general formula ABC(CN). When B=
Fe2+ and C= Co3+ the compound is diamagnetic because both ions are in
their low spin, non-magnetic
non magnetic state. By illuminating with red light however it
is possible to induce an electron transfer in which Fe2+ is changed
to low spin Fe3+, with one unpaired electron,
and Co3+ to high spin Co2+ with three unpaired electrons:
Fe2+-C-N-Co3+ - Fe3+-C-N-Co2+
A schematic drawing of the light induced transformation is shown in the
previous slide. The material orders as a bulk ferrimagnet below 50 K. If the
irradiation is performed below this temperature we observe a transition to
bulk magnetic
g
order induced by
y light.
g Therefore these materials can be
considered as magnetic switches operated by light. t is also possible to
perform the opposite transition by irradiating the Fe3+-Co2+ pairs with blue
light: the electron is back transferred from cobalt to iron and the system
reverts to the diamagnetic state.
Chapter
Ch
t VIII
VIII.
Mixed-valence
Mixed
valence compounds.
The phenomenon of mixed valency.
Spin dependent delocalization
Spin-dependent
delocalization-double
double
exchange- classical and quantummechanical description (Anderson’s
theory). Robin and Day classification of
mixed-valence
i d l
compounds.
d IIntervalence
t
l
light absorption (light induced electron
t
transfer).
f ) Magnetic
M
ti properties.
ti
MIXED VALENCE COMPOUNDS
Unpaired electron is delocalized - metal ions,
(let say, A and B) are in a different oxidation states:
states
+1
d n – d n+1
Unpaired electron can be found at each site, so that two
configurations
fi
ti
are equivalent
i l t iin energy:
d An d Bn 1 and
d An 1 d Bn
Simplest case:
electron delocalized over spinless
p
metal sites, d0– d 1 cluster.
Assume that a and b are the orbitals of the electron at the
corresponding sites, the energies are equal. The trapped
states are unstable, the kinetic energy and Coulomb
attraction to the alien site promote the electron transfer
process with the rate t which can be associated with the
transfer integral.
SPLITTING
S h
Scheme
off the
th molecular
l
l orbitals
bit l – stationary
t ti
delocalized states:
u
a
2|t|
g
b
Providing t 0 :
1
g a b bonding
g orbital
2
1
u a b antibondin g orbital
2
g and u symbols of parity
Bonding and antibonding orbitals are of the opposite parity,
so the light absorption and emission processes in electric
dipole approximations are allowed.
Intervalence absorption bands- one of the main
manifestation of the mixed valence
valence.
Crucial role of the vibronic interaction
STRUCTURE OF THE FERROMAGNETIC
[Ni2(napy)4Br2]+
t
(
(napy)=
)
SELECTED EXAMPLES OF MIXEDMIXED-VALENCE
COMPOUNDS
Molecular structure of a
binuclear Fe(II)-Fe(III)
mixed-valence dimer
Molecular structure of a
binuclear Mn(III)-Mn(IV)
mixed-valence dimer
FULL--SYMMETRIC (“BREATHING”) VIBRATION
FULL
z
Octahedral ML6
complex:
coordinate
system and
enumeration of
the ligands
3
4
5
x
2
1
6
R>R0
Q
y
R0- equiliblium M-L
distance
Xi, Yi, Zi – displacements
di l
t
R=R0
R<R0
1
X 1 X 4 Y2 Y5 Z 3 Z 6
6
VIBRONIC PIEPHO-KRAUZS-SCHATZ (PKS) MODEL
FOR MIXED –VALENCE
VALENCE COMPOUNDS
N
N
N
N
N
N
N
N
A
B
N
N
Q>0
q>0
N
N
N
N
N
N
N
A
N
N
N
N
N
B
Q=0
N q=0
N
Out-ofOutof-phase mode of
a dimeric moiety:
N
N
N
N
A
N
N
N
N
N
B
N
N
Full-symmetric
“breathing” local
vibrations of
independent
fragments AN6
and
d BN6 off a
dimeric unit
Q<0
N q<0
Q
1
2
QA QB
POTENTIAL SURFACES OF A MIXED-VALENCE DIMERROBIN AND DAY CLASSIFICATION,, PKS MODEL
2
Q 2Q 2 t 2
2
pseudo
pse do Jahn - Teller vibronic
ibronic coupling,
co pling t transfer integral,
integral
frequency of the active vibration
U Q
Q
Class A
strongly localized
t
2
1
Q
Class B
partially delocalized
t
2
1
Q
Class C
fully delocalized
t
2
1
INTERVALENCE ABSRPTIONABSRPTION- MAIN
MANIFESTATION OF MIXED VALENCE
a
adiabatic
c potenttial
Blue arrowsarrows-intervalence transitions of the light absorption
8
8
8
6
6
6
4
4
4
2
2
2
0
0
0
-2
-2
-2
-4
4
-4
4
-4
4
-4
0
q
4
uncoupled sites,
full localization
-4
0
q
4
Intermediate vibronic
coupling, moderate
electron
l t
ttransfer,
f
partial delocalization
-4
0
q
4
Weak vibronic coupling
or/and strong electron
t
transfer,
f
full delocalization
CONCEPT OF THE DOUBLE EXCHANGE
Magnetic exchange (HDVV) –coupling of localized spins:
SA
SB
Double exchange interaction –coupling of two
localized magnetic moments having spins S0 through
an itinerant (traveling) electron that can travel forth
and back between two magnetic centers.
traveling electron
e
Spin core A
S0
S0
Spin core
B
ELECTRON TRANSFER
BETWEEN LOCALIZED SPINS
Let us consider now the general case of a
mixed valence dimer dn-dn+1.
The first problem in question is how the magnetic moment of
th metal
the
t l iions affect
ff t the
th electron
l t
transfer.
t
f It happens
h
that,
th t in
i
this case, electron transfer is spin-dependent. The main
features of the phenomenon can be understood in the
framework of the classical spin model developed by
Anderson and Hasegawa: Phys.
Phys Rev
Rev.10 (1955) p.675
Rev.10
p 675.
As distinguished from a quantum spin, which can be
oriented in the space
p
in 2S+1 directions,, a classical spin
p
represents the infinite spin limit for which
all the directions in the space are allowed
(illustration-next Slide).
QUANTUM AND “CLASSICAL” SPINS
m=+1/2
m=+1
m=5/2
m=3/2
m=1/2
m=0
m=-1/2
m=-3/2
m=-1/2
1/2
S = 1/2
m=-1
1
S=1
m=-5/2
5/2
S = 5/2
S =
Quantum spin-distinct
spin
effect of spatial quantization,
classical spin (high spin) all directions in the space are
allowed
ll
d - smallll angles
l b
between
t
th
the vectors
t
S.
HUND’S
HUND
S RULE
Let us consider a high-spin state (Hund´s configuration)
for the dn+1 ions.
ions From the classical point of view,
view that
means that the extra electron lines up its spin, parallel
to the spin core
core, taking thus the gain in energy from
the intraatomic exchange.
s
S0
extra electron
spin core
Intraatomic exchange –ferromagnetic!
INTERMEDIATE SPIN VALUES IN
CLASSICAL SPIN REPRESENTATION
s
S0
A
B
=0
Smax?S 0
int er m ed iat e
=
Smin
i ?
Illustration for the different spin values in a pair of ions
with delocalized extra electron.
COMMENT TO THE CLASSICAL PICTURE
For the MV dimer,, the full spin
p of the system
y
can
take 2S+1 values with S comprised between
Smax = 2So+1/2 and Smin = 1/2.
In the classical limit, So >> 1/2, so that
Smax ≈ 2So and Smin ≈ 0.
0
These two extreme correspond to
parallel and antiparallel
orientations of the spin cores while the
intermediate spin values are to be correlated
with intermediate angles between the spin cores.
FINAL RESULT IN CLASSICAL
APPROXIMATION (P.W.Anderson)
SA
S
/2
S
t S t
2S0
SB
The main physical result:
Rate of the electron transfer proves to be spindependent and increases with the increase of the full
spin of the systems
High-spin states are stabilized more strongf
ferromagnetic
ti effect
ff t
QUANTUM PICTURE OF THE SPINSPINDEPENDENT ELECTRON TRANSFER
-DOUBLE EXCHANGE
a
b
a
b
a
b
a
b
1
A*B
1
1
AB*
1
Scheme of the electron jumps between two localized
configurations.
g
Quantum-mechanical result for the
double exchange
g splitting
p
g ((Anderson(Anderson-Hasegawa):
g
)
S 1/ 2
E S t
2 So 1
DOUBLE-EXCHANGE SPLITTING IN A PAIR
DOUBLEOF S=1/2 IONS WITH A DELOCALIZED
ELECTRON
S=3/2
SA=1, SB=1/2
(S=1/2, 3/2)
A*B
localized
u
S=1/2 g
t 2t
S=1/2 u
S=3/2
S
/ g
delocalized
SA=1/2, SB=1
(S=1/2, 3/2)
AB*
localized
Ferromagnetic
g
g
ground state,, rule of p
parity
y alternation.
DOUBLE EXHANGE SPLITTING IN MIXEDMIXEDVALENCE DIMERS WITH THE INCREASING SPINS
d0-d
d1
d1-d
d2
d2-d
d3
d3-d
d4
d4-d
d5
2/3t
1/2t
2/5t
S=9/2
Classical Spin Limit
(continuous spectrum)
+t
2t
-t
t
S=1/2
S=3/2
S=5/2
S=7/2
S0=0
S0=1/2
S0=1
S0=3/2
S0=2
S0=
General splitting is the same for all dimers,
ferromagnetic effect,
effect density of spin-levels
spin levels
increases with the increase of spin core
Energy in
n the units of tran
nsfer para
ameter
MIXED-VALENCE DIMER Fe(II)
MIXEDFe(II)--Fe(III),
SPIN--DEPENDENT DELOCALIZATION
SPIN
E/t
u
1
The main
observations:
g
u
g
u
0
g
u
g
u
g
-1
0
2
4
6
spin multiplicity
8
10
2S+1
1) Ferromagnetic
effect of the double
exchange
2)) Rule
ueo
of a
alternation
te at o
for the even and
odd spin-levels
VIBRONIC LOCALIZATION IN THE D1-D2 MV DIMER
(t and v in vibrational energy units)
(S=3/2,1/2)
S
3/2
1/2
t
2t
1/2 3/2
1/2
3/2
1/2
3/2
t=1
t=1
ν=0
v=0
t=1
t=1
ν=2
v=2
t=1
t=1
ν=4
v=4
1/2
3/2
-3
-2
-1
0
1
Ferromagneticfully delocalized
2
3
-4
-2
0
2
4 -6
Weakly
ferromagneticpartially localized
-4
-2
0
2
q
4
6
Paramagneticfully localized
MORE COMPLICATED MIXEDMIXED-VALENCE SYSTEMS
(only to mention)
t
Mixed-valence
polyoxometalates,
18 Sites
Sites, 1
1- 8 moving
electrons
t'
Fulleride anion C602-,
two moving electrons
STRUCTURE AND THE MODEL CALCULATION
Network
et o o
of tthe
e
exchange and
electron transfer
parameters
Model
system
y
Antiferromagnetic
effect of delocalizationS=0- ground state
QUESTIONS FOR THE EXAMINATION
1. Magnetic substances, the main kinds of the magnetic behavior.
2. Spin, spatial quantization, quantum and classical pictures.
3 Zeeman interaction for a free spin and Zeeman splitting.
3.
splitting
4. Boltzman distribution, partition function for a spin in an external
magnetic field.
5. Magnetic susceptibility , magnetization-basic equations, Brillouin
function, saturation in a strong field.
6. Curie low, effective magnetic moments, transition metals, rarerare
earth ions.
7. Electron paramagnetic resonance, quantum and classical pictures
of the phenomenon,
phenomenon principle of detection
detection.
9. Quantum numbers of a free atom, angular momentum opertors.
10. Spin-orbit
p
splitting,
p
g Zeeman splitting
p
g for a LSJ term, vector
model, g-factors of a free atom.
11. EPR and magnetic susceptibility for the LSJ states, magnetism
of rare
rare-earth
earth ions: Gd(III),
Gd(III) Eu(II),
Eu(II) Sm(II),
Sm(II) Eu(III).
Eu(III)
12. Splitting of the free ion terms in a crystal field, a qualitative
picture and group-theoretical rules.
13. Parameter of the cubic field (Dq), spectrochemical series, lowsymmetry complexes
complexes, electrond and holes in crystal field
formalism.
14. Matrices of spin and orbital momentum operators. Matrix
elements
l
t off spin-orbital
i
bit l iinteraction,
t
ti
evaluation
l ti off th
the g-factors
f t
for
f
Cu(II) ion in tetragonal symmetry.
15. Anisotropy of g-factors and magnetic susceptibility, reduction of
the orbital contribution by the crystal field.
16. EPR in anisotropic complexes, angular dependences of
g-factors,, analysis
g
y of the powder
p
samples,
p , shape
p of the EPR lines
in different symmetries.
17. Covalence and the orbital reduction factor, manifestation in EPR.
18 Zero-field
18.
Zero field splitting physical mechanism
mechanism, qualitative picture
( group-theoretical consideration).
19. Zero-field splitting, concept of spin-Hamiltonian, energy levels
f Ni(II) and
for
dC
Cr(III)
(III) complexes.
l
20. Zero –field
field splitting, energy levels, EPR, allowed and forbidden
transitions, anisotropy, advantages of multifrequency EPR.
21. Exchange interaction, Pauli principle, spin-functions, exchange
and Coulomb integrals
integrals.
22. The concept of spin-Hamiltonian for the exchange, spincoupling- classical picture, eigen-problem.
23. Cooper(II) dimers, magnetic moments.
24. Heisenberg-Dirac- Van Vleck model for the exchange, multielectron p
problem of exchange,
g ,p
polynuclear
y
systems.
y
25. Trimeric magnetic clusters, spin coupling scheme, Kambe’s
approach.
26 Energy levels and EPR of dimeric and trimeric clusters
26.
clusters.
27. Two kinds of tetrameric systems, exchange Hamiltonians, spin
coupling.
28. Phenomenon of single molecule magnetism, physical principlesquantum tunneling and relaxation, possibilities of application as
g density
y storage
g units in computers
p
,p
physical
y
requirements.
q
high
29. Mixed valence, electron delocalization, vibronic models,
intervalence optical absorption.
30. Double exchange, classical spin theory, ferromagnetic effect.
Vibronic interactions
interactions.





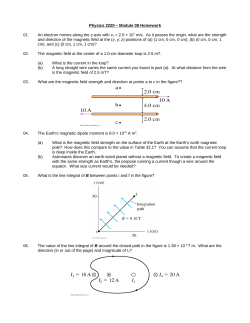



© Copyright 2025