
Organic Chemistry
Content ORG: Organic and Green Chemistry Tittle Authors Paper ID 309 SYNTHESIS AND CHARACTERIZATION OF STAR-SHAPED PYRENYL TRIAZATRUXENES 166 OXIDATION OF THIOL INTO DISULFIDE USING POLYSTYRENE-SUPPORTED (DIACETOXYIODO)BENZENE 353 330 217 201 434 273 Thanachart Techajaroonjit, Vinich Promarak, Paitoon Rashatasakhon Watanya Krailat, Preecha Phuwapraisirisan, Tirayut Vilaivan, Mongkol Sukwattanasinitt, Sumrit Wacharasindhu Weerawat Sripet, SYNTHESIS OF HEXAPHENYLBENZENE DERIVATIVES CONTAINING IMINE MOIETY Mongkol Sukwattanasinit, AS FLUORESCENCE TURN-ON PROBE FOR Sumrit Wacharasindhu THE DETECTION OF METAL IONS SYNTHESIS OF TRUXENE DERIVATIVES Chomchanok Wongsilarat, WITH DIPYRENYLCARBAZOLE PENDANTS Vinich Promarak, Paitoon Rashatasakhon Kananat Naksomboon, CYCLIC ALKYL SULFIDE DIETHER Krittaphat Wongma, Phairat DERIVATIVE AS AN ELECTRON DONOR Phiriyawirut, Tienthong FOR ZIEGLER-NATTA CATALYST IN Thongpanchang OLEFIN POLYMERIZATION ALPHA-BROMINATION OF KETONE USING Tipakorn Sangrawee, HEXABROMOACETONE Warinthorn Chavasiri Natthida Maneechandra, FRIEDEL–CRAFTS BENZYLATION OF TOLUENE USING NdCl3 IMPREGNATED ON Warinthorn Chavasiri ALUMINIUM PILLARED MONTMORILLONITE SYNTHESIS AND CHARACTERIZATION OF Patthira Sumsalee, Pages 239-242 243-246 247-251 252-255 256-259 260-262 263-266 267-270 Paper ID 209 172 342 Tittle ISOINDIGO DERIVATIVES AS MOLECULAR DONORS FOR ORGANIC PHOTOVOLTAICS SYNTHESIS TOWARDS N- Authors Visit Waewsungnoen, Vinich Promarak Chalupat Jindakun, HETEROAROMATIC THENA DERIVATIVE: Thanawon Chaisantikulwat, INVESTIGATION ON [4+2]-CYCLOADDITION Jakapun Soponpong, Kulvadee Dolsophon, OF HIGHLY REACTIVE PYRIDYNE Tienthong Thongpanchang CHEMOSELECTIVE REDUCTIONS OF Thamonwan Angkuratipakorn, NITROBENZENE USING Pawinna Chanthawong, CHITOSAN-COATED METAL AS A Jirada Singkhonrat CATALYST SYNTHESIS AND CHARACTERIZATION OF Chittranuch Pengsawad, CYANURIC ACID SUBSTITUTED 1,8Mongkol Sukwattanasinitt, NAPHTHALIMIDES Paitoon Rashatasakhon Pages 271-274 275-278 279-282 239 SYNTHESIS AND CHARACTERIZATION OF STAR-SHAPED PYRENYL TRIAZATRUXENES Thanachart Techajaroonjit1, Vinich Promarak2, Paitoon Rashatasakhon1* 1 Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand School of Chemistry and Center of Excellence for Innovation in Chemistry, Institute of Science, Suranaree University of Technology, Muang District, Nakorn Ratchasima, 30000, Thailand 2 * Email: paitoon.r@chula.ac.th Abstract: Triazatruxene derivatives have received much attention in the research field of organic lightemitting diode (OLED) due to their unique optical properties and excellent thermal stabilities. In this research project, we designed and synthesized two molecules of symmetrical triazatruxene derivatives containing 2-ethyl hexyl or benzyl groups. The triazatruxene has been synthesized from the Br2catalyzed cyclotrimerization of indole. A sequential debromination-bromination thus provides a tribromo triazatruxene core with a complete regioselectivity. Alkylation of the N-H group and Suzuki crosscoupling with pyrene-1-boronic acid give rise to the target compounds in good yields. After the characterization by 1H-NMR, 13C-NMR, IR, and mass-spectrometry, their photophysical properties are investigated by UV-Vis and fluorescent spectrophotometry. In this paper, a series of novel star-shaped 3,8,13-substituted triindoles were reported. They were synthesized via a bromine-catalyzed cyclotrimerisation of indole (Figure 1). The substitution by pyrene and alkyl groups were later performed in order to obtained the expected compounds. The characterizations and photophysical property investigations were also reported. Br N H N H HN 1. Introduction Br Research on organic light-emitting diodes (OLEDs) has been continuously developed during the last few decades. OLED display has thus become a theme of interest due to its advantages such as properties for large area, flexible, lightweight, and energy efficient optoelectronics [1,2]. π-Conjugated materials are extensively investigated and explored for OLEDs because of their potential in the creation of cost-effective, power-efficient, and flexible electronic devices [35]. Nowadays aromatic cores with a large π-orbital area are used as hole-transporting materials. These molecules have a strong tendency to assemble in highly ordered organizations caused by stacking, which paves the way for a favorable overlap of πorbitals [6-8]. Carbazole derivatives are well-known holetransporting units because of their electrondonating capabilities associated with the nitrogen atom [9,10]. From this reason, they were used as important building blocks for OLED materials. One of the electron-rich carbazole derivatives, 10, 15dihydro-5H-dihydro-5H-diindolo [3,2-a:3’,2’c]carbazole or triindole, has became a newly famous compound. It possesses aromatic surface with three facile points for attachment of side chains: the three indolic NH groups in the 5-, 10and 15-positions. Triindole core was synthesized by cyclocondensation of either indolin-2-one or indole through halogenations [11]. NH N H NH N H Figure 1. Br2-catalyzed cyclotrimerisation of indole 2. Experimental 2.1 Materials All reagents were purchased from Aldrich, Fluka and used without further purification. 2.2 Measurements All 1H NMR spectras were recorded on Varian Mercury 400 MHz NMR spectrometer (Varian, USA) using CDCl3 and acetone-d6. 13C NMR spectras were recorded at 100 MHz on Bruker 400 MHz NMR spectrometer using the same solvent. Mass spectra were recorded on a Microflex (Bruker MALDI-TOF mass spectrometer Daltonics) using doublyrecrystallized α-cyano-4hydroxy cinnamic acid (CCA) as a matrix. Absorption spectras were measured by a Shimadzu UV-2550 UV-Vis spectrophotometer. Fluorescence spectras were obtained from an Agilent technologies Cary Eclipse spectrofluorometer. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 240 2.3 Synthesis 2.3.1 10,15-Dihydro-5H-dihydro-5Hdiindolo[3,2-a:3’,2’-c]carbazole (4): A mixture of indole 3 (2 g, 17 mmol) and CH3CN (5 mL) was stirred at room temperature and solution of Br2 (1.3 mL, 51 mmol) in CH3CN (15 mL) was added over 5 min. The mixture was stirred overnight, and the resulting dark-green solid was filtered and washed with acetonitrile (250 mL). At this point, without further purification, the crude product (3 g) was mixed with Et3N (8.4 mL, 60.46 mmol), HCOOH (2.28 mL, 60.46 mmol) and 10% Pd/C (400 mg, 0.36 mmol) in MeOH (50 mL), and the resulting mixture was heated for 30 min under reflux. The mixture was filtered through Celite, and the filtrate was diluted with CH2Cl2, washed with aqueous HCl (10%) and dried (Na2SO4), and the solvent was evaporated under reduced pressure to give brown solid (2 g). The crude product was dissolved in methanol, adsorbed onto silica gel and purified by chromatography (ethyl acetate/n-hexane, 15:85) to give pure 4 as a pale-yellow solid (0.16 g, 16%). 1H NMR (400 MHz, acetone) δ 11.16 (s, 3H), 8.57 (d, J = 7.5 Hz, 3H), 7.74 (d, J = 7.8 Hz, 3H), 7.35 (dt, J = 22.8, 7.2 Hz, 6H). 2.3.2 3,8,13-Tribromo-10,15-dihydro-5Hdiindolo [3,2-a:3’,2’-c]carbazole (5): A solution of N-bromosuccinimide (NBS) (0.28 g, 1.55 mmol) in dimethylformamide (2 mL) was added dropwise to a mixture of 4 (0.17 g, 0.5 mmol) in acetone (10 mL) at 0 °C. The mixture was slowly warmed to room temperature and stirred for an additional 30 min before it was poured into water. Then, the organic phase was separated and dried over anhydrous Na2SO4. After the solvent was evaporated, the crude product was purified by column chromatography using hexane/acetone (8:2) as the eluent to afford 5 as a pale white solid (0.22 g, 76%). 1H NMR (400 MHz, acetone) δ 11.34 (s, 3H), 8.39 (d, J = 8.3 Hz, 3H), 7.85 (s, 3H), 7.45 (d, J = 6.8 Hz, 3H). 2.3.3 5,10,15-Triethylhexyl-10,15-dihydro-5Hdiindolo[3,2-a:3’,2’-c]carbazole (6): A mixture of 5 (0.15 g, 0.25 mmol) and KOH (0.28 g, 5 mmol) was stirred at room temperature, then a solution of ethylhexylbromide (0.27 mL, 1.5 mmol) was added slowly, the mixture was stirred overnight. The mixture was poured into water and extracted with EtOAc. The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure., the crude product was purified by chromatography (EtOAc:n-hexane, 5:95) to give the compound 6 as a yellow solid (0.22 g, 95%). 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.5 Hz, 3H), 7.52 (s, 3H), 7.46 (d, J = 8.6 Hz, 3H), 4.40 (s, 6H), 1.79 (s, 3H), 1.10 – 0.44 (m, 42H). 2.3.4. 5,10,15-Tribenzyl-10,15-dihydro-5Hdiindolo[3,2-a:3’,2’-c]carbazole (7): A mixture of 5 (0.15 g, 0.25 mmol), KOH (0.28 g, 5 mmol), and [CH3(CH2)3]4N(HSO4) (0.0083 g, 0.025 mmol) was heated to reflux in acetone (10 mL). Benzyl bromide (0.2 mL, 1.68 mmol) was then added and the mixture was stirred for 3 h. The mixture was diluted with CH2Cl2, washed with 10% aqueous HCl and with saturated aqueous NaCl solution, and dried (Na2SO4), the solvent was then evaporated. The residue was triturated with hexanes to give 7 as a white solid (0.21 g, 98%). 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 3H), 7.89 (d, J = 7.0 Hz, 3H), 7.74 (d, J = 8.5 Hz, 3H), 7.47 (m, 9H), 7.10 (d, J = 7.2 Hz, 6H), 5.96 (s, 6H). 2.3.5 5,10,15-Triethylhexyl-3,8,13-tri(pyren-1yl)-10,15-dihydro-5H-diindolo[3,2-a:3',2'c]carbazole (1): To a degassed (N2) solution of 6 (0.09 g, 0.1 mmol) and Pd(PPh3)4 catalyst (0.012 g, 0.01 mmol) in toluene (5 mL), then pyreneboronic acid (0.1476 g, 0.6 mmol) and 2 M aqueous K2CO3 solution (1 mL) were added via syringe. The reaction mixture was stirred at 70 oC for 48 h. After cooling, the product was extracted with CH2Cl2, washed with water, and dried over anhydrous Na2SO4. The solvent was evaporated, affording the crude mixture. The crude product was purified by column chromatography (CH2Cl2:n-hexane, 1:9) to give the compound 1 as a yellow solid (83.1 mg, 65%). 1H NMR (400 MHz, CDCl3) δ 8.47 (d, J = 9.2 Hz, 3H), 8.35 – 7.88 (m, 30H), 7.67 (s, 3H), 5.09 (s, 6H), 2.28 (d, J = 12.1 Hz, 3H), 1.70 (s, 6H), 1.41 (s, 3H), 1.2 – 0.55 (m, 39H). 13C NMR (100 MHz, CDCl3) δ 150.2, 141.6, 136.5, 131.8, 131.31, 131.25, 130.6, 129.8, 129.1, 129.0, 128.3, 127.7, 127.6, 127.5, 126.5, 126.2, 126.0, 125.34, 125.25, 124.9, 124.8, 122.7, 122.2, 113.8, 51.1, 38.6, 34.5, 33.9, 28.5, 23.3, 14.0, 10.6. MALDITOF MS (m/z): calcd: (1282.693 [C96H87N3]); found: (1281.797 [M+]). 2.3.6 5,10,15-Tribenzyl-3,8,13-tri(pyren-1-yl)10,15-dihydro-5H-diindolo[3,2-a:3',2'-c]carbazole (2): To a degassed (N2) solution of 7 (85.2 mg, 0.1 mmol) and Pd(PPh3)4 catalyst (11.6 mg, 0.01 mmol) in toluene (5 mL), then pyreneboronic acid (14.8 mg, 0.6 mmol) and 2 M aqueous K2CO3 solution (1 mL) were added via syringe. The reaction mixture was stirred at 70 oC for 48 h. After cooling, the product was extracted with CH2Cl2, washed with water, and dried over anhydrous Na2SO4. The solvent was evaporated, affording the crude mixture. The crude product was purified by column chromatography (CH2Cl2:n-hexane, 2:8) to give the compound 2 as a yellow solid (0.06 g, 49%). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 7.9 Hz, 3H), 7.90 (m, 30H), 7.70 (d, J = 9.2 Hz, 3H), 7.47 (s, 3H), 7.38 (d, J = 6.6 Hz, 6H), 7.28 (d, J = 7.2 Hz, 6H), 6.04 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 141.8, 140.3, 138.2, 138.1, 136.4, 131.6, 131.1, 130.4, 129.3, 128.6, 128.1, 127.7, 127.5, 127.4, 127.3, 126.8, 126.0, 125.5, 125.02, 124.98, 124.7, 124.6, 123.4, 122.5, 121.7, 113.2, 103.5, 51.5. MALDI-TOF MS (m/z): calcd: (1216.459 [C93H57N3]); found: (1215.485 [M+]). Pure and Applied Chemistry International Conference 2015 (PACCON2015) 241 3.2 Photophysical properties of compound 1 and 2 The investigation of absorption properties of 1 and 2 in CHCl3 revealed a similar absorption wavelength and molar extinction coefficient (Table 1 and Figure 2). However, the emission spectra of compound 1 appeared at a slightly longer wavelength with higher quantum efficiency than 2. This might attribute to the steric hindrance of the three 2-ethylhexyl groups that enhances the solubility of the compound in organic solvents and prevents aggregation-caused fluorescent quenching. 3. Results and Discussion 3.1 Synthesis of 1 and 2 (Scheme 1) Our synthesis of triazatruxene relied on the cyclotrimerization of indole, which was catalyzed by Br2. The formation of dimer and several oligomers of indole could be the main cause for the poor efficiency of this reaction. Upon the reductive de-bromination using trimethylammonium formate, the regioselective tribromination using NBS provide tribromoazatruxene 5 in 76%. The following alkylation of the N-H group gave rise to 6 and 7. Finally, the Suzuki cross-coupling pyrene1-boronic acid afforded target compounds 1 and 2 in good yields. Table 1: Photophysical properties of 1 and 2 Cpd. 1 Absorption λmax log ε (M-1cm-1) (nm) 344 4.52 Emission λmax ΦFa (nm) 483 0.52 2 351 472 HN 1) Br2, MeCN, RT, 1d N H N H 2) Et3N, HCOOH, Pd/C, MeOH, reflux, 30min 3 NH quinine sulfate in 0.1 M H2SO4 (ΦF = 0.54) was used as the standard. acetone, RT, 30 min NBS, DMF R Br N 0.61 a 4 Br 4.88 HN Br Br a or b N R N H N R Br Br 6 or 7 B(OH)2 NH 5 Pd(PPh3)4 K2CO3, toluene, 70 oC, 2d Figure 2. Normalized absorption and emission spectra of 1 and 2 in CHCl3 4. Conclusions R N N R N R R1 = 1 R2 = In this research, two new symmetrical pyrenyl triazatruxene derivatives were successfully synthesized via Br2-catalyzed cyclotrimerization of indole and Suzuki cross-coupling with pyrene-1boronic acid in good yields. The substitution of the -NH position by 2-ethylhexyl and benzyl groups could prevent the aggregation by pi-stacking. These compounds were well soluble in organic solvents and exhibited high quantum efficiencies. Acknowledgements 2 Scheme 1. Synthesis of 1 and 2: Reagent and conditions: (a) 2-ethyl hexylbromide, KOH, DMSO, RT, overnight; (b) benzyl bromide, KOH, Bu4N∙HSO4, acetone, reflux, 3h. This work was financially supported by the Faculty of Science, Chulalongkorn University. The instruments were partly sponsored by the National Research University Project of Thailand, Office of the Higher Education Commission (AM1006A) Pure and Applied Chemistry International Conference 2015 (PACCON2015) 242 and the Thai Government Stimulus Package 2 (TKK2555, SP2). References [1] Weiss, D.S. and Abkowitz, M., 2010, Chem. Rev., 110, 1, 479-526. [2] Forrest, S. R. and Thompson, M.E., 2007. Chem. Rev., 107, 4, 923-925. [3] Gustafsson, G., Cao, Y., Treacy, G.M., Klavetter, F., Colaneri, N., and Heeger, A. J., 1992, Nature, 357, 477-479. [4] Martin, R.E. and Diederich, F., 1999, Angew. Chem. Int. Ed., 38, 10, 1350-1377. [5] Kraft, A., Grimsdale, A.C., and Holmes, A.B., 1998, Angew. Chem. Int. Ed., 37, 4, 402-428. [6] Zang, L., Che, Y., and Moore, J.S., 2008. Acc. Chem. Res., 41, 12, 1596-1608. [7] Schenning, A.P.H. J. and Meijer, E.W., 2005, Chem. Comm., 26, 3245-3258. [8] Wu, J., Pisula, W., and Muellen, K., 2007. Chem. Rev., 107, 3, 718-747. Zhang, Y., Wada, T., and Sasabe, H., 1998. J. Mater. Chem., 8, 809-828. [10] Grazulevicius, J.V., Strohriegl, P., Pielichowski, J., and Pielichowski, K., 2003, Prog. Polym. Sci., 28, 9, 1297-1353. [11] Franceschin, M., Ginnari-Satriani, L., Alvino, A., Ortaggi, G., and Bianco, A., 2010, Eur. J. Org. Chem., 1, 134-141. [9] Pure and Applied Chemistry International Conference 2015 (PACCON2015) 243 OXIDATION OF THIOL INTO DISULFIDE USING POLYSTYRENESUPPORTED (DIACETOXYIODO)BENZENE Watanya Krailat1, Preecha Phuwapraisirisan2, Tirayut Vilaivan2, Mongkol Sukwattanasinitt3 and Sumrit Wacharasindhu3* 1 Program of Petrochemistry and Polymer Science, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand 2 Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand 3 Nanotec-CU Center of Excellence on Food and Agriculture, Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand * E-mail: sumrit.w@chula.ac.th, Tel: +66-2218-7634, Fax : +66-2218-7598 Abstract: Disulfides are important compounds in biological and chemical processes which can be prepared by the oxidation of thiols. For this work, we have developed a new preparative method for the disulfide using inexpensive, recyclable, and relatively non-toxic polymer-supported (diacetoxyiodo)benzene (PS-DIB) as the oxidant under mild conditions. PS-DIB, a yellow powder, was prepared from iodination of commercially available polystyrene (MW=35,000) followed by oxidation with either NaBO3.4H2O or Ac2O/H2O2 in 43% and 70% yields, respectively. The structure of the prepared PS-DIB was confirmed by FTIR, which exhibited two strong bands at 1639 and 1712 cm-1 corresponding to the carbonyl group. The loading of the (diacetoxyiodo)phenyl group on polystyrene was determined by iodometric titration and calculated to be 1.39 mmol/g. The reaction conditions were optimized for solvent, time and equivalent of PS-DIB using 4-chlorothiophenol as substrate and the yields were determined using HPLC. We discovered that, under the optimized condition (iPrOH as solvent, 1 equivalent of PS-DIB, open air, 1 h, room temperature) for 4-chlorothiophenol, the corresponding disulfide product was obtained in 79% yield following chromatography. This methodology could be extended to a variety of thiols, giving the corresponding disulfides in good yields. 1. Introduction Disulfides are important compounds that are widely used in both biological and chemical processes. They were used as reagents to stabilize polypeptide secondary structure in peptide and protein synthesis.[1] Moreover, disulfides are important reagents for vulcanization in rubbers and elastomers.[2] Typical synthesis of disulfides involves the conversion of the corresponding thiols via the use of strong oxidizing agent or metal catalyst such as cerium(IV)salts, transition metal oxides,[3] H2O2,[4] 2,6-dicarboxy pyridinium chlorochromate,[5] halogens[6] and heterogeneous permanganate.[7] Those reagents are not only toxic but also expensive in some cases.[8] Therefore, the development of thiol oxidation under mild and environmental friendly condition remain a challenging. Recently, hypervalent iodine reagents were introduced to organic synthesis as oxidizing agents due to their low cost, mild and highly selective oxidizing properties, environmental friendly character and commercial availability.[9] We recently reported the use of a hypervalent iodine, diacetoxyiodo(benzene) for oxidation of thiols into disulfide in good yields.[2] Even though the reaction condition is mild and may be carried out in open flask condition, the difficulty to remove the iodobenzene byproduct from the desired disulfide makes the reuse of the diacetoxyiodo(benzene) reagent cumbersome. To overcome this problem, the hypervalent iodine is bound onto a polymeric support. The desired product can be obtained by simple filtration and the polymersupporting reagent could be regenerated and recycled. Therefore, in this work we reported the synthesis of polystyrene-supported diacetoxyiodo(benzene) (PSDIB) and its use as an oxidizing agent for a variety of thiols into disulfides. 2. Materials and Methods 2.1. Materials All starting materials were obtained from SigmaAldrich, Fluka (Switzerland) or Merck (Germany) and used without further purification. Analytical thin-layer chromatography (TLC) was performed on Kieselgel F254 pre-coated plastic TLC plates from EM Science. Visualization was performed with a 254 nm ultraviolet lamp. Gel column chromatography was carried out with silica gel (60, 230-400 mesh) from ICN Silitech. The 1H and 13C NMR spectra were recorded on a Varian 400 or Bruker 400 in CDCl3. 2.2 Synthesis 2.1 Synthesis of poly (4-iodostyrene) (PS-I) To a mixture of nitrobenzene (50 mL), CCl4 (10 mL), H2SO4 (50%, 9 mL) were added polystyrene (4 g, 38.25 mmol), I2 (4.5 g, (17.75 mmol), and I2O5 (1.79 g, 5.36 mmol) at 90 °C and the reaction was stirred at that temperature for 72 h. After the reaction was complete, MeOH 400 mL was added into the reaction mixture and the precipitate was collected by filtration and washed with MeOH and dried. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 244 2.2 Synthesis of polystyrene-supported (diacetoxyiodo)benzene (PS-DIB) Method A [9] A solution of poly (4-iodostyrene) (5.2 g), AcOH (113 mL), DCE (7 mL), and TfOH (135.66 mmol) was stirred at 40°C. Then, NaBO3.4H2O (135.7 mmol) was added slowly within 2 h. After evaporation of DCE and AcOH, H2O 40 mL was added into the reaction. The precipitate was filtered, washed with MeOH and dried. Method B [10] A solution of 30% H2O2 (27 mL, 895.01 mmol) was added dropwise into Ac2O (97 mL, 1030.91 mmol) and stirred at 40 °C for 4 h. Then, poly(4-iodostyrene) (5.2 g) was added into the reaction mixture and stirred overnight. Et2O 100 mL was then added into the reaction. The precipitate was filtrated, washed with MeOH and dried under vacuum. 2.3 Iodometric titration The loading of the reagent was determined by iodometric titration. A mixture of PS-DIB (0.031 g), KI (0.250 g), deionized water (12.5 mL), H2SO4 (6 N, 1.25 mL), and CHCl3 (1.25 mL) were added in 50-mL flask and stirred for 4 h. The reaction mixture was titrated with 0.1 N sodium thiosulfate using starch solution as indicator. white solid: 1H NMR (CDCl3, 400 MHz) δ 8.40 (d, J = 8.1 Hz, 2H), 7.60-7.49 (m, 4H), 7.04 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 159.0, 159.0, 149.6, 149.6, 137.4,137.4, 121.1, 121.1, 119.7, 119.7; IR (neat, cm-1) 3168, 3098, 1604, 1570, 1487, 1438; IR (neat, cm-1) 3046, 2987, 1574, 1556, 1444, 1414; LRMS (ESI) calcd for C10H9N2S2, 221.01; found, 221.01. 3. Results and Discussion 3.1 Synthesis and characterization of polymersupported (diacetoxyiodo)benzene PS-DIB To prepare PS-DIB, the commercially available polystyrene (MW=35,000) was iodinated by I2/I2O5 in the mixture of CCl4/nitrobenzene solution under acidic condition as presented in Scheme 1. This resulted in the formation of iodo-polystyrene (PS-I) as yellowish green solid (Figure 1 (middle)). Then, the desired PSDIB was obtained by oxidation with either the use of Ac2O/H2O2 or NaBO3.H2O as shown in Scheme 1. After the reaction, the precipitate was collected by filtration, washed with MeOH, and dried under vacuum. Both oxidation procedures gave yellow powder of PS-DIB as shown in Figure 1 (right). A 2.4 Synthesis of disulfides (1-3) using PS-DIB 1,2-Bis(4-chlorophenyl)disulfane (1): 4Chlorothiophenol (50.0 mg, 0.346 mmol) and PS-DIB (249 mg, 0.346 mmol) were mixed in i-PrOH (5 mL) in a round-bottomed flask with a magnetic stir bar and stirred for 60 min. The solvent was removed by rotary evaporation and the crude product was purified by silica gel chromatography (100% hexane) to afford 1,2bis(4-chlorophenyl)disulfane (39 mg, 0.173 mmol, 79%) as a yellow solid; 1H NMR (CDCl3, 400 Hz) δ 7.40 (d, J = 8.7 Hz, 4H, Ar-H), 7.27 (d, J = 8.7 Hz, 4H, Ar-H); 13C NMR (CDCl3, 100 Hz) δ 135.2, 133.7, 129.4, 129.3; IR (neat, cm-1) 3079, 2925, 1470, 1385. 6,6’-Disulfanediyldihexan-1-ol (2): 6-Mercapto-1hexanol (50.76 µL, 0.372 mmol) and PS-DIB (669 mg, 0.930 mmol) were mixed in i-PrOH (5 mL) in a roundbottomed flask with a magnetic stir bar and stirred for 60 min. The solvent was removed by rotary evaporation and the crude product was purified by silica gel chromatography (80% EtOAC/hexane) to afford 6,6’-disulfanediyldihexan-1-ol (51 mg, 0.186 mmol, quantitative) as a yellow solid; 1H NMR (CDCl3, 400 Hz) δ 3.58 (t, J = 6.5 Hz, 4H, CH2-OH), 2.62 (t, J = 8.0 Hz, 4H, CH2-S), 1.62 (m, 4H, -CH2-), 1.51 (m, 4H, -CH2-), 1.34 (m, 8H, -CH2-); 13CNMR (CDCl3, 100 Hz) δ 62.8, 62.8, 39.1, 39.1, 32.6, 32.6, 29.1, 29.1, 28.2, 28.2, 25.4, 25.4; IR (neat, cm-1) 3346, 2934, 2859, 1465, 1053 2,2’-Dithiodipyridine (3): 2-Mercaptopyridine (50.0 mg, 0.450 mmol) and PS-DIB (324 mg, 0.450 mmol) were mixed in i-PrOH (5 mL) in a round-bottomed flask with a magnetic stir bar and stirred for 60 min. The solvent was removed by rotary evaporation and the crude product was purified by silica gel chromatography (30% EtOAC/hexane) to afford of 2,2’-dithiodipyridine (37.2 mg 0.225 mmol, 75%) as a Ac2O/30%H2O2 40oC, 4 h I2, I2O5, CCl4 50% H2SO4, nitrobenzene 90 oC, 72 h I PS-I NaBO3.H2O/TfOH/AcOH/DCE AcO I OAc 40oC, 4 h PS-DIB B Scheme 1. Synthesis of PS-DIB from method A and B PS PS-I PS-DIB Figure 1. Color and appearance of PS (left), PS-I (middle) and PS-DIB (right) The structure of PS-DIB was confirmed by FTIR spectroscopy by comparision with unmodified polystyrene (PS) and iodopolystyrene (PS-I) as shown in Figure 2. The PS-I showed the reduction of C-H (aromatic) streching peak at 3017 cm-1. These results suggested the succesful iodination of the benzene ring in polystyrene. Moreover, the structure of polystyrenesupported (diacetoxyiodo) benzene (PS-DIB) was confirmed by the existence of peaks at 1639 and 1712 cm-1 which can be assigned as C=O streching in the diacetoxy moiety. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 245 PS 3.3 Utilization of PS-DIB for oxidization of thiols PS-I 3.3.1 Optimization study PS-DIB Figure 2. Overlay of FT-IR spectra of PS, PS-I and PSDIB 3.2 Determination of the loading of functional group of PS-DIB + K2SO4 + I2 +2HOAc + 2KI + H2SO4 AcO I I OAc 2NaI + Na2S4O6 2Na2S2O3 + I2 Scheme 2. Iodometric titration Table 1: The loading of functional group Loading of diacetoxyiodo group Washed (mmol) in PS-DIB time Method A Method B 2.13 1.87 1st nd 2 2.28 1.61 3rd 2.36 1.19 4th 1.90 1.19 5th 1.41 1.19 The loading of the diacetoxyiodo group on polystyrene was determined by iodometric titration and the reaction equation was presented in Scheme 2. PSDIB and KI were dissolved in a solution of chloroform and sulfuric acid to produce I2 via oxidation reaction. Then, it was titrated with sodium thiosulfate with starch solution as the indicator. The results from both PS-DIB from method A and method B were depicted in Table 1. In order to remove the excess reagents from the prepared PS-DIB and to test the stability of the diacetoxyiodo group bounded to the polymer, we washed the PS-DIB for 5 times and the loading after each washing was evaluated as shown in Table1. We found that the amount of the loading of the (diacetoxyiodo)phenyl group on the polystyrene prepared from Method A decreased continually after washing with MeOH while the PS-DIB obtained from Method B remained constant after the third wash. In Method A, the oxidation with Ac2O and H2O2 may not be very efficient. The high loading observed before the methanol rinse is perhaps due to the remaining unreacted H2O2, which decreased eventually after the washing. Based on this discovery, the oxidation with NaBO3.H2O (Method B) was used as a standard method for the preparation of PS-DIB throughout this work. Next, we investigated the PS-DIB reactivity for the conversion of 4-chlorothiophenol (1) into 1,2-bis(4chlorophenyl)disulfane (1a). During the optimization, the amount of PS-DIB and reaction time were varied and the results were summarized in Table 2. When the reaction was carried out in the presence of 0.7 equivalent of PS-DIB for 60 min, 72% yield of the disulfide was obtained (Table 2, entry 1). Increasing the amount of PS-DIB from 0.7 to 1.1 quivalent resulted in the formation of the desired disulfide in 77% yield (Table 2, entry 2). However, if we conducted the reaction only for 5 minute, the reaction efficiency decreased (Table 2, entry 3). Therefore, the utilization of 1.1 equivalent of PS-DIB for 60 minute was considered as the optimized condition for oxidization of thiols. Table 2: Oxidation of 4-chlorothiophenol with PS-DIB Entry 1 2 3 SH Time (min) 60 60 5 PS-DIB Equivalent 0.7 1.1 1.1 Cl PS-DIB S i -PrOH, rt Cl % yield (HPLC) 72 77 67 (1) Cl S (1a) 3.3.2 Substrate scope With the optimized condition in hand, we next explored the scope of PS-DIB oxidization. A variety of thiols containing alkyl or heterocyclic substituent were subjected to the optimized condition and the results were shown in Table 3. 6-Mercapto-1-hexanol (2) was treated with PS-DIB in i-PrOH and the corresponding disulfide (2a) was generated in quantitative yield after column chromatography (Table 3, entry1). For heterocyclic substrate, 2-mercaptopyridine (3) was oxidize into the corresponding disulfide (3a) in 75% yield. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 246 Table 3: Synthesis of disulfides from thiols References [1] PS-DIB (1.1 eq.) R-SH (1-3) i-PrOH, rt, 60 min R-S-S-R (1a-3a) R= Alkyl, Aromatic, Heterocyclic %Yielda Thiols Entry SH 79 1 Cl 2b HO 3 a b (1) SH (2) Quantitative 75 N SH (3) Thurow, S., Pereira, V.A., Martinez, D.M., Alves, D., Perin, G., Jacob, R.G. and Lenardão, E.J., 2011, Tetrahedron Lett., 52, 640-643. [2] Zhdankin, V. Z., 2009. ARKIVOC, (i), 1-62. [3] Akdag, A., Webb, T. and Worley, S. D., 2006. Tetrahedron Lett., 47, 3509–3510. [4] He, Y., Hang, D. and Lu, M., 2012. Phosphorus Sulfur Silicon Relat. Elem., 187, 9, 1118-1124. [5] Tajbakhsh, M., Hosseinzadeh, R. and Shakoori, A., 2004, Tetrahedron Lett., 45, 1889-1893. [6] Hashmat Ali, M., McDermott, M., 2002, Tetrahedron Lett., 43, 6271–6273. [7] Walters, M. A., Chaparro, J., Siddiqui, T., Williams, F., Ulku, C. and Rheingold, A.L., 2006, Inorg. Chim. Acta., 359, 3996–4000. [8] Attri, P., Gupta, S. and Kumar, R., Green Chem. Lett. Rev. 2012, 33-42. [9] Chen, F.-E., Xie, B., Zhang, P., Zhao, J.-F., Wang, H. and Zhao, L., 2007, Synlett, 4, 619-622. [10] Hossain, M. D., Kitamura, T., 2006, Synthesis, 8, 12531256. isolated yield 2.5 eq. PS-DIB were used. 4. Conclusions We successfully synthesized polystyrene-supported diacetoxyiodo(benzene) from commercially available polystyrene in good yield with high loading of (diacetoxyiodo)phenyl group. The polymer-supported reagent was able to oxidize a variety of thiols into the corresponding disulfides in good to excellent yields. The reaction is not only efficient but also proceeds smoothly under mild conditions in open flasks using relatively less toxic i-PrOH solvent. Acknowledgements This work is financially supported by the Thailand Research Fund (RSA5780055) and Nanotechnology Center (NANOTEC), NSTDA, Ministry of Science and Technology, Thailand, through its program of Center of Excellence Network. This work is part of the Project for Establishment of Comprehensive Center for Innovative Food, Health Products and Agri-culture supported by the Thai Government Stimulus Package 2 (TKK2555, SP2), the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission (AM1006A-56), the Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University (RES560530126-AM) and the 90th anniversary of Chulalongkorn University Fund (Ratchadaphiseksomphot Endowment Fund). Pure and Applied Chemistry International Conference 2015 (PACCON2015) 247 SYNTHESIS OF HEXAPHENYLBENZENE DERIVATIVES CONTAINING IMINE MOIETY AS FLUORESCENCE TURN-ON PROBE FOR THE DETECTION OF METAL IONS Weerawat Sripet, Mongkol Sukwattanasinit, Sumrit Wacharasindhu* Nanotec-CU Center of Excellence on Food and Agriculture, Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand E-mail: sumrit.w@chula.ac.th, Tel. +662 2187634, Fax. +66 22187598 Abstract: Currently, the design and synthesis of new fluorescent chemosensors for the efficient detection of metal ions is one of the most important research topics in environmental chemistry and biology. In this work, we select hexaphenylbenzene (HPB) as a fluorophore and imine moiety as a receptor for metal ions. The HPB containing salicylaldehyde group (HPB-SW1) is successfully obtained via Diels-Alder reaction between 2hydroxy-5(phenylethynyl)benzaldehyde and tetraphenylcyclopentadienone in 51% yield. The target fluorophores [HPB-SW (2-4)] are received from condensation of HPB-SW1 with the corresponding amines such as 2-aminophenol, n-propylamine and ethanolamine in 74-89% yields. These compounds are characterized by 1H-NMR, 13C-NMR and HRMS. The addition of Al3+ to sensor HPB-SW2 induces strong blue fluorescence emission while the HPB-SW3 and 4 show selective turn-on fluorescence toward Zn2+ ion.The detection limit of HPB-SW4 is calculated to be 10.46 ppb for Zn2+ which is lower than drinking water permission concentration by world health organization (WHO). 1. Introduction The development of fluorescent chemosensor for metal ions has received considerable attention [1] due to their biological and environment important roles. Among various metal ions, zinc ion (Zn2+) has attracted a great deal of attention ascribing to its biological significance. Zinc ion plays significant role in various fundamental biological processes, such as gene transcription and DNA binding or recognition. Also, excessive amount of zinc in human cause many severe diseases such as Alzheimer’s disease, Friedreich’s ataxia and Parkinson’s disease [2]. On the other hand, aluminium is widely used in many applications such as textile industry, medicines, paper industry and food additive. An excess amount of aluminium in human body not only damages the central nervous system but also causes various diseases such as Alzheimer’s, Parkinson’s and breast cancer [2]. The traditional analytical methods, such as atomic absorption spectroscopy, inductively coupled plasma mass spectroscopy and electrochemical analysis, have been reported for the trace-quantity determination of metal ions. But, most of those methods are expensive and time-consuming in practice. In comparison, fluorescence spectroscopy is widely used because of its high sensitivity and ability to perform on-site analysis making fluorescence approach superior to other analytical methods. Therefore, it is of great importance to develop highly specific fluorescence turn-on probe for the detection of Zn2+ and Al3+ in aqueous media. Most reported flurophores, however, exhibit aggregation causing quenching (ACQ) phenomenon leading to low quantum efficiency in solid state or at high water content [3]. Until recently, there are reports on antiACQ flurophore molecules such as tetraphenylethylene, hexaphenylsilole and hexaphenylbenzene showing aggregation induced emission effect [4]. These allow such flurophores to perform the detection of metal ions in solid state or aqueous solution. Herein we report the preparation of novel fluorescent chemosensors based on hexaphenylbenzene derivatives HPB-SW (2-4). These compounds were attached with different imine moieties as metal ion receptor as shown in Figure 1. HPB-SW2 and HPB-SW4 demonstrate the specific and sensitive fluorescence turn-on properties with Al3+ and Zn2+ ions, respectively OH N OH HPB-SW2 N N OH OH OH HPB-SW3 HPB-SW4 Figure1. Structures of HPB-SW2, HPB-SW3 and HPB-SW4 2. Materials and Methods 2.1 Instrument and reagents All reagents were purchased from SigmaAldrich, Fluka® (Switzerland) or Merck® (Germany) and used without further purification. Analytical thinlayer chromatography (TLC) was performed on Kieselgel F-254 pre-coated plastic TLC plates from EM Science. Visualization was performed with a 254 nm ultraviolet lamp. Column chromatography was carried out with silica gel (60, 230-400 mesh) from ICN Silitech. The 1H and 13C NMR spectra were obtained on a Varian Mercury NMR spectrometer, which operated at 400 MHz for 1H and 100 MHz for 13C nuclei (Varian Company, CA, USA). Mass spectra were recorded on a Microflex MALDI-TOF Pure and Applied Chemistry International Conference 2015 (PACCON2015) 248 mass spectrometer (Bruker Daltonics) using doubly recrystallized α-cyano-4-hydroxy cinnamic acid (CCA) and dithranol as a matrix. Absorption spectra were measured by a Varian Cary 50 UV-Vis spectrophotometer. Fluorescence spectra were obtained from a Varian Cary Eclipse spectrofluorometer. 2.2 Synthesis Synthesis of 5-iodosalicylaldehyde (1) Salicylaldehyde (0.870 mL, 8.189 mmol) was dissolved in dichloromethane, and followed by addition of iodine monochloride (0.692 mL, 13.228 mmol) at 0 oC to room temperature. The reaction mixture was left overnight. After the reaction was completed, the reaction mixture was extracted with CH2Cl2 and the organic solution was washed with Na2S2O3, respectively. The organic layer was dried over anhydrous magnesium sulfate and solvent was removed by evaporator then recrystallized from hot hexane temperature to afford the white solid compound 1 ( 65%). 1H NMR (400 MHz, CDCl3) δ 10.95 (s, 1H), 9.83 (s, 1H), 7.85 (s, 1H), 7.77 (d, J = 8.5 Hz, 1H), 6.80 (d, J = 8.5 Hz, 1H). 13C NMR (101MHz, CDCl3) δ ppm 195.3, 161.3, 145.5, 142.0, 122.7, 120.1, 80.4 . Synthesis of 2-hydroxy-5-(phenylethynyl)benzaldehyde (2) 5-Iodosalicylaldehyde (1.80 g, 7.258 mmol) was mixed with Pd(PPh3)2Cl2 (0.457g, 0.653 mmol), CuI (0.124 g, 0.653mmol) and PPh3 (0.085 g, 0.326 mmol) in round bottom flask under N2 atmosphere. After that, THF and TEA were added and kept stirred for 15 min. Then, phenylacetylene (2.38 mL, 21.77 mmol) was gradually added. The mixture was left overnight. The rotary evaporator was used to evaporate solvent from the mixture. The residue was purified by column chromatography on silica gel (10% EtOAc in hexane) to give compound 2 (79% yield). 1H NMR (400 MHz, DMSO) δ (ppm) 11.17 (s, 1H), 10.25 (s, 1H), 7.78 (s, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.58 – 7.47 (m, 2H), 7.47 – 7.34 (m, 3H), 7.04 (d, J = 8.5 Hz, 1H). 13 C NMR (101 MHz, CDCl3) δ (ppm) 196.5, 162.8, 161.9, 140.3, 137.3, 132.0, 128.9, 123.4, 120.8, 118.3, 115.7, 89.7, 87.6. Synthesis of compound HPB-SW1 A solution of compound 2 (0.300 g, 1.34 mmol) and tetraphenylcyclopenta-2,4-dienone (0.468 g 0.218mmol) in 3 mL of diphenylether was refluxed for overnight under nitrogen atmosphere. The dark-brown mixture was diluted with dichloromethane (2 mL), poured in methanol (50 mL) and stirred; the off white precipitate was filtered and recrystallized from methanol to afford compound HPB-SW1 (51% yield). 1 H NMR (400 MHz, DMSO) δ (ppm) 10.29 (s, 1H), 9.89 (s, 1H), 7.09 (d, J = 1.8 Hz, 1H), 7.00 (d, J = 8.5 Hz, 1H), 6.95 – 6.72 (m, 25H), 6.45 (d, J = 8.5 Hz, 1H). 13C NMR (101 MHz, DMSO) δ (ppm) 191.13, 157.9, 140.1, 140.1, 139.9, 139.9, 138.7, 138.5, 131.6, 131.4, 130.8, 130.8, 130.7, 126.6, 126.4, 125.3, 125.2, 120.5, 115.5, 47.2. General procedure for the synthesis of HPB-SW (24) A clear solution of compound HPB-SW1 and amine compounds in dry THF: EtOH (2:3) was stirred at 65 °C. After 24 hrs, the reaction mixture turned turbid. The reaction mixture was concentrated under the reduced pressure and dry ethanol was poured into it, solid appeared. The solid was filtered and recrystallized from methanol to afford the compound HPB-SW (2-4) Synthesis of compound HPB-SW2 The compound HPB-SW1 (0.100 g, 0.173 mmol) reacted with 2-aminophenol (0.0283 g, 0.260 mmol) to afford the light yellow solid compound HPB-SW2 (74% yield); 1H NMR (400 MHz, CDCl3) δ (ppm) 8.20 (s, 1H), 7.17 (m, 1H), 6.99 (d, J = 7.4 Hz, 2H), 6.86 (d, J = 15.0 Hz, 26H), 6.67 (d, J = 7.5 Hz, 1H), 6.55 (d, J = 8.3 Hz, 1H), 6.31 (d, J = 7.6 Hz, 1H). 13C NMR (101MHz, DMSO) δ (ppm) 161.3, 158.1, 150.9, 140.3, 140.1, 140.0, 140.0, 139.0, 135.4, 134.8, 134.6, 130.9, 130.8, 130.7, 127.9, 126.6, 126.5, 126.4, 125.3, 125.2, 119.5, 119.4, 117.8, 116.5, 115.5, 114.9. HRMS (ESI); m/z calcd for C49H35NO2 + H+: 670.2668 [M+H+]: found, 670.2740. Synthesis of compound HPB-SW3 The compound HPB-SW1 (0.150g, 0.259mmol) reacted with propan-1-amine (0.023g, 0.388mmol) to afford the light yellow solid compound HPB-SW3 (88% yield); 1H NMR (400 MHz, CDCl3) δ (ppm) 7.78 (s, 1H), 6.76 (s, 26H), 6.66 (d, J = 8.6 Hz, 1H), 6.54 (s, 1H), 6.36 (d, J = 8.0 Hz, 1H), 3.33 (d, J = 5.8 Hz, 1H), 1.13 (d, J = 6.2 Hz, 6H). 13C NMR (101MHz, CDCl3) δ (ppm) 161.9, 140.6, 140.4, 140.3, 139.2, 134.1, 131.3, 126.9, 126.7, 126.6, 125.2, 125.2, 24.0. HRMS (ESI); m/z calcd for C46H37NO + H+: 620.2875 [M+H+]: found, 620.2958. Synthesis of compound HPB-SW4 The compound HPB-SW1 (0.100 g, 0.173 mmol) reacted with 2-aminoethanol (0.0158 g, 0.260 mmol) to afford the light yellow solid compound HPB-SW4 (89% yield);1H NMR (400 MHz, DMSO) δ (ppm) 13.26 (s, 1H), 8.10 (s, 1H), 6.99 – 6.73 (m, Pure and Applied Chemistry International Conference 2015 (PACCON2015) 249 27H), 6.31 (d, J = 8.6 Hz, 1H), 4.70 (d, J = 4.9 Hz, 1H), 3.54 (t, J = 4.5, 2H), 3.50 (t, J = 4.5 Hz, 2H). 13C NMR (101 MHz, DMSO) δ (ppm) 165.7, 158.3, 140.3, 140.1, 140.1, 139.9, 139.9, 139.1, 134.7, 133.8, 130.8, 130.2, 126.5, 126.4, 125.3, 125.3, 116.9, 114.8, 60.8, 60.5. HRMS (ESI); m/z calcd for C45H35NO2 + H+: 622.2668 [M+H+]: found, 622.2745. 3. Results and Discussion The preparations of compounds HPB-SW2, HPB-SW3 and HPB-SW4 are depicted in Scheme 1. Initially, HPB-SW1 were obtained from [4+2] cycloaddition reaction between tetraphenylcyclopenta2,4-dienone (3) and diphenylacetylene (2) in 51% yield as a white solid after recrystallized from methanol. Then the target HPB-SW2, HPB-SW3 and HPB-SW4 were synthesized from HPB-SW1 via the imine formation with the corresponding amine such as 2-aminophenol, n-propylamine and 2-aminoethanol in 74-89 % yields, respectively. To confirm the structure of the prepared compounds, the 1H NMR, 13C NMR, and HRMS were performed. The mass analysis data correspond to all prepared compounds. 1H NMR analysis of HPB-SW1 and HPB-SW4 (Figure2.) confirms the success of imine formation showing that phenolic group shift downfield from 10.31 to 13.28 ppm and the aldehyde peak at 9.91 ppm disappeared. Also the new peaks at 8.17, 4.74, 3.56 and 3.51 ppm are corresponding to imine, primary alcohol and methylene protons, respectively. 3.1 Effect of water content on photophysical properties The HPB derivatives are well-known flurophores that can exhibit aggregation induced emission (AIE) due to their restricted intramolecular rotations behaviors (RIR) [4]. Therefore, we began the investigation of the AIE effect on our flurophore HPB-SW2, HPB-SW3 and HPB-SW4 by studying the relationship between the amount of water in THF solution toward the relative fluorescence intensity (I/I0) (Figure 3.). In case of HPB-SW2, it showed an obvious fluorescence enhancement upon increasing the ratio of water from 0 to 60%, suggesting the AIE effect (Figure1.). However, the addition of water higher than 60% led to a decrease in the emission intensity. This result may be attributed to the low solubility of HPB-SW2 in the solvent mixture, leading to a decrease in the number of emissive molecules per unit volume. In this case, intramolecular rotations are restricted in aqueous media because of the formation of aggregates, which block the nonradiative channels and populate the radiative excitons, thereby making the molecule emissive in their aggregated state [5]. OH OH CHO CHO I-Cl/CH2Cl2 0 0C PdCl2(PPh3)2, CuI,PPh3 OH TEA. rt, N2, overnigth to rt,overnigth CHO I 1 2 O 2 R NH2 OH Diphenylether, 260 oC CHO -CO dry THF:EtOH (2:3) Stirr. 65 oC HPB-SW (2-4) Figure3. Fluorescence intensity of HPB-SW2 (λem 524 nm), HPB-SW3 (λem 460 nm) and HPB-SW4 (λem 470 nm) 20 µM in THF: H2O mixtures at different water contents. All compounds were excitated at 280 nm. HPB-SW1 Scheme 1. Synthetic route of compound HPB-SW2, HPB-SW3 and HPB-SW4 Figure2. Overlay 1H NMR spectra of HPB-SW1 (top) and HPB-SW4 (bottom) in d6-DMSO On the other hand, the fluorescence intensity of HPB-SW3 and HPB-SW4, gradually decreased as the fraction of THF in the mixed THF/water solution decrease (100:0 to 10:90 v/v) (Figure 3.). The strong emission of HPB-SW3 and HPB-SW4 in 100%THF may be attributed to non-aggregated molecules. In contrast, the decreasing fluorescence intensity of HPB-SW3 and HPB-SW4 along with a slight red shift from 455 to 500 nm was observed. To the best of our knowledge, these are the first example of HPB derivatives that have no AIE effect. We hypothesized that the imine linkage possesses imine isomerization along with the free rotation from alkyl side chain resulting to the non-radiactive decay [7]. This behavior not only reduces the fluorescence intensity but also suppress the aggregation. Therefore, flurophore HPB- Pure and Applied Chemistry International Conference 2015 (PACCON2015) 250 SW3 and HPB-SW4 should be suitable for the design of “turn-on” fluorescence sensor. 3.2 Fluorescent chemosensor To demonstrate the sensing ability of compound HPB-SW2, HPB-SW3 and HPB-SW4, we firstly evaluated the relative fluorescence intensity (I/I0) of those probe toward various metal ions such as Cr3+, Al3+, Zn2+, Ni2+, Co2+, Ag+, Ba2+, Pb2+, Ca2+, Cu2+, Cd2+, Hg2+, Mg2+, Na+, K+, Fe3+ and Fe2+ in pure water. When excited at 280 nm, the HPB-SW2 alone displayed a weak emission band at 453 nm in THF/water (4:6 v/v) solution. Upon the addition of 10 equivalent of each metal ions, only Al3+ induced a 1.3fold enhancement in emission intensity while other metal ions showed either no or slight fluorescence intensity change (Figure 4a.inset). Similarly, the HPBSW3 and HPB-SW4 alone displayed a very weak emission band at 453 and 457 nm respectively with excitation at 280 nm in THF/water (1:1 v/v) solution. Upon the addition of 10 equivalents of each metal ion, only Zn2+ induced large fluorescence enhancement of HPB-SW3 and HPB-SW4 to 15-fold and 25-fold respectively. There was no observable change upon the addition other metal ions (Figure 4). Importantly, visual evidence of the specific and strong turn-on fluorescence of the probe HPB-SW4 with Zn2+ were observed as strong blue emission of solution under the black light as seen in figure 4b. From these observation, HPB-SW4 probe prove to be the best turn-on florescence sensor for detection of Zn2+ showing higher selectivity and sensitivity in the comparison with HPB-SW2 and HPB-SW3. Moreover, this result suggests that the hydroxyl moiety and flexible alkyl side chain in the imine receptor facilitate the binding efficiency with Zn2+ ion. 280 nm. The insert bar graph represents the change of the relative emission intensity of compound HPBSW2 in THF: H2O (4:6, v/v) λex 280 nm. 4b. Photographs show selectivity of HPB-SW4 (50 μM) upon mixing with different other metal cations (10eq) in THF:H2O (1:1, v/v) under illumination by a UV lamp. To quantify the sensing ability of HPB-SW4 sensor, the fluorescence titration with Zn2+ was carried out (Figure5). The emission intensities of HPB-SW4 gradually increased as the concentration of Zn2+ increased. Fluorescence intensity increased linearly up to addition of 30 equiv of Zn2+ indicating that the it might not be a simple 1:1 complex. Thus we hypothesized that after the formation of complex HPBSW4-Zn2+, the excess Zn2+ also induced the formation of aggregates in solution. However, we need to further investigate their morphorlogy in order to confirm our hypothesis. Fianlly, the detection limit of HPB-SW4 as a fluorescence chemosensor for analysis of Zn2+ was found to be 0.160 µM which is far below the World Health Organization guideline (76 µM) [6]. Figure5. Change in the fluorescence spectra of HPBSW3 (20 μM) upon a gradual increase in the concentration of Zn2+ in THF: H2O (1:1, v/v) 4. Conclusions Figure 4a. Bar graph representing the change of the relative emission intensity of compounds HPB-SW3 and HPB-SW4 (20μM) upon mixing with different other metal ions (10eq.) in THF: H2O (1:1, v/v) λex We have successfully synthesized novel flurogenic probe HPB-SW2, HPB-SW3 and HPBSW4 based on hexaphenylbenzene. The HPB-SW2 containing hydroxyphenyl imine demonstrated turn-on fluorescence for Al3+ with high selectivity. Moreover, the HPB-SW4 composed of 1-imineethanol moiety, exhibited an excellent selectivity and sensitivity toward Zn2+ ions as turn-on fluorescence mode. These sensors might have the potential application for the detection of Al3+ and Zn2+ in water quality monitoring. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 251 Acknowledgements This work is financially supported by the Thailand Research Fund (RSA5780055) and nanotechnology Center (NANOTEC), NSTDA, Ministry of Science and Technology, Thailand, through its program of Center of Excellence Network. This work is part of the Project for Establishment of Comprehensive Center for Innovative Food, Health Products and Agriculture supported by the Thai Government Stimulus Package 2 (TKK2555, SP2), the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission (AM1006A-56) and the Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University (RES560530126-AM). References [1] Chen, X., Pradhan, T., Wang, F., Kim, J and Yoon J., 2012, Chem Rev., 112, 19101-956. [2] Choi, Y.W., Park, G.J., Na, Y.J., Jo, H.Y., Lee ,S.A., You G.R and Kim, C., 2014, Sensors and Actuators B: Chemical., 194, 343-352. [3] Hong, Y, Lam, J.W.Y. and Tang, B.Z., 2011, Chem. Soc. Rev., 40, 5361–5388. [4] Kumar, M., Vij, V. and Bhalla, V., 2012, Langmuir., 28, 12417−12421. [5] Dong, S., Li, Z. and Qin, J. 2009, J. Phys. Chem. B., 113, 434-441. [6] Kumar, Y. K., King, P. and Prasad, V. S. K. R., 2006, Chem. Eng. J., 124, 63-70. [7] Jiasheng, W., Weimin, L., Jiechao, Ge., Hongyan, Z. and Pengfei W. 2011, Chem. Soc. Rev., 40, 3483–3495. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 252 SYNTHESIS OF TRUXENE DERIVATIVES WITH DIPYRENYLCARBAZOLE PENDANTS Chomchanok Wongsilarat1, Vinich Promarak2, Paitoon Rashatasakhon3* 1 Program in Petrochemistry and Polymer Science, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand School of Chemistry and Center of Excellence for Innovation in Chemistry, Institute of Science, Suranaree University of Technology, Muang District, Nakorn Ratchasima, 30000, Thailand 3 Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330 Thailand 2 * E-mail: paitoon.r@chula.ac.th, Tel. +6622817620 Abstract: Derivatives of truxene exhibited excellent photophysical properties along with good thermal and electrochemical stabilities suitable for application in optoelectronic devices. In this paper, we reported the synthesis and characterization of truxene derivatives with different numbers of dipyrenylcarbazole substituents. The truxene core is readily accessible via the dehydro-cyclotrimerization of hydrocinnamic acid. The solubility in organic solvents is enhanced when the methylene units of truxene are substituted by n-butyl groups. The dipyrenylcarbazole pendant is prepared from iodination of carbazole followed by a Suzuki coupling with the commercially available pyrene-1boronic acid. The Cu-catalyzed C-N coupling between the iodinated truxene core and dipyrenylcarbazole thus provides the target 1 and 2 which are characterized by 1 H NMR, 13C NMR and MALDI-TOF mass spectroscopy. The photophysical properties of both compounds are examined by UV-visible and fluorescence spectroscopy. 1. Introduction Organic light–emitting devices (OLEDs) have been more interesting because of their applications in flat panel displays. There have been extensive studies on layered organic electroluminescent (EL) devices to enhance device performances [1-2]. High efficiency and good durability are generally important for practical applications. One of the main reasons for the degradation of the OLED device is the morphological change in the amorphous organic layers, especially of the hole transport layer, caused by Joule heating during device operation. In order to solve this issue, it is necessary to develop amorphous materials with a high glass transition temperature (Tg). Therefore, different synthetic approaches have been studied to produce novel high Tg hole-transport materials (HTMs) to generate thermally stable OLEDs. Recent development in emissive materials has focused on the blue electroluminescence (EL) as a number of new fluorescent blue light-emitting materials, such as triphenylfluoranthene, fluorene, triarylamine and pyrene derivatives have been reported. However, pyrene derivatives are more attractive to their strong fluorescence, high quantum efficiency, carrier mobility, and hole-transporting ability [3, 8]. On the other hand, it is well-known that carbazole is a good hole-transporting and electroluminescent group, and many LED materials contain carbazole moieties as the key constructing block. The exceptional holetransporting ability of the carbazole-containing derivatives is attributed to the electron donating capabilities of carbazole moieties. Furthermore, the chemical and thermal stabilities of carbazole derivatives are extremely high, and the carbazole ring can be easily functionalized at the 3-, 6-, and 9positions [4-7, 9-10]. On the other hand, truxene or 10,15-dihydro-5H-diindenol[1, 2-a:10, 20-c]-fluorene is a planar heptacyclic polyarene. It can be formally regarded to as a C3-symmetrically fused fluorene trimer. Because its unique three-dimensional topology which could be comfortably functionalized by different substituents at C-2, -7, -12 positions and at C5, -10, -15 positions, truxene has been thoroughly developed as an attractive building block and starting material for numerous functional organic materials such as OLEDs, fluorescence sensor, organic solar cells as well as large pi-conjugation dendrimer macromolecular [9-13]. For these reasons, we designed and synthesized two novel compounds by a combination of 3,6-dipyrenylcarbazole units with truxene core and hypothesized that they could serve as hole-transporting materials in OLEDs. To emphasize our progress of this project, the synthesis and characterization of these compounds are reported herein. 2. Materials and Methods 2.1 Chemical and instruments All reagents were purchased from Aldrich, Fluka and used without further purification. All 1H NMR spectra were recorded on Varian Mercury 400 MHz NMR spectrometer (Varian, USA). 13C NMR spectra were recorded at 100 MHz on Bruker NMR spectrometer. Mass spectra were recorded on a Microflex MALDITOF mass spectrometer (Bruker Daltonics) using doubly recrystallized α-cyano-4-hydroxy cinnamic acid (CCA) as a matrix. Absorption spectra were measured by a Varian Cary 50 UV-Vis spectrophotometer. Fluorescence spectra were obtained from a Varian Cary Eclipse spectrofluorometer. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 253 2.2 Synthesis 2.2.1 3,6-Diiodo-9H-carbazole (4) To a stirred solution of carbazole (5.0 g, 29.9 mmol) in acetic acid was added potassium iodide (6.6 g, 39.8 mmol). Then, potassium iodate (9.6 g, 44.9 mmol) was added in small portions over a period of 5 min and the resulting mixture was refluxed for 20 min. The reaction was allowed to cool to room temperature and diluted with EtOAc. The combined organic layer was dried over MgSO4 filtered, and concentrated under reduced pressure to give a brown solid residue. The crude product was purified by recrystallization from acetone and hexane to yield 4 as light brown crystals (12.3 g, 98%).1H NMR (400 MHz, DMSO) δ 11.56 (s, 1H), 8.57 (s, 2H), 7.65 (d, J = 7.6 Hz, 2H), 7.35 (d, J = 7.6 Hz, 2H) ppm. 2.2.2 3,6-Di(pyren-1-yl)-9H-carbazole (5) A mixture of 4 (1.4 g, 3.3 mmol), pyrene-1-boronic acid (2.0 g, 8.1 mmol), Pd(PPh3)4 , 2M K2CO3 aqueous solution was heated refluxing in THF conditions for 24 h. After the reaction was cooled to room temperature, the resulting brown solution was extracted with CH2Cl2. The combined organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography using hexane: CH2Cl2 (3:1) as the eluent to yield 5 as pale green solid (1.23 g, 66%). 1H NMR (400 MHz, DMSO) δ 11.69 (s, 1H), 8.49 (s, 2H), 8.36 (d, J = 7.9 Hz, 2H), 8.30 – 8.11 (m, 14H), 8.06 (t, J = 7.6 Hz, 2H), 7.77 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 8.3 Hz, 2H) ppm. MALDI-TOF MS: C44H25N found 567.004 ([M]+ calcd: 567.198) 2.2.3 10,15 dihyhro-5H-diindeno[1,2-a:1’,2’-c] fluorene (Truxene) (6) [11,12] 3-Phenylpropionic acid (10.02 g, 66.72 mmol) was mixed with polyphosphoric acid (50 g) and heated at 60 °C for 30-40 min in nitrogen atmosphere. Water (5 mL) was then added to the reaction and temperature was raised to 160 °C for 3 h. After the reaction was cooled to room temperature, the mixture was poured into ice water and grey powder was filtered off under suction and washed with water. The residue was recrystallized from toluene to yield 6 as light-yellow power (11.12 g, 49%). 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 7.1 Hz, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.40 (d, J = 7.3 Hz, 1H), 4.22 (s, 2H) ppm. 2.2.4 5,5,10,10,15,15-Hexabutyl-truxene (7) A solution of truxene (6) (1.00 g, 2.92 mmol) in DMF (50 mL) at 0 °C under nitrogen, NaH (1.19 g, 29.8 mmol) was added and the solution was allowed to warm to room temperature and stirred for 30 min, then n-butyl bromide (3.2 mL) was added for 24 h. The mixture was poured into water and extracted with EtOAc. The combined organic layer was dried over MgSO4, filtered, and concentrated under reduce pressure. The crude product was purified by silica gel column chromatography using hexane as the eluent to yield 7 as white solid (1.48 g, 75%). 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 7.3 Hz, 1H), 7.46 (m, 2H), 7.38 (m, 2H), 3.04 – 2.91 (m, 2H), 2.15 – 2.04 (m, 2H), 0.96 – 0.79 (m, 4H), 0.62 – 0.36 (m, 10H) ppm. 2.2.5 5,5,10,10,15,15-Hexabutyl-2-iodo-truxene (8) 5,5,10,10,15,15-Hexabutyl-2,7-diiodo-truxene (9) A mixture of truxene (7) and solvent (CH3COOH:H2SO4:H2O:CCl4) = 100:5:20:8) was heated to 40 °C. After adding HIO3 and I2 to the mixture, the mixture was heated to 80 °C and stirred for 4 h at this temperature. After the reaction was completed, the mixture was cooled to room temperature and filtered off under suction, washed with water. Then the residue refluxed in methanol for 2h and followed by cooling to room temperature, filtered off under suction, and white power was obtained (68% yield 8). 1H NMR (400 MHz, CDCl3) δ 8.4-8.3 (m, 2H), 8.14 - 8.04 (d, J = 8.5 Hz, 1H), 7.77 (s, 1H), 7.75 - 7.68 (d, J = 8.2 Hz, 1H), 7.49 - 7.42 (m, 2H), 7.43 - 7.32 (m, 4H), 3.05 - 2.78 (m, 6H), 2.1 1.95 (m, 6H), 0.98 - 0.8 (m, 12H), 0.62 - 0.34 (m, 30H) ppm. (64% yield 9) 1H NMR (400 MHz, CDCl3) δ 8.48.3 (m, 1H), 8.14-7.97 (d, J = 8.5 Hz, 2H), 7.77 (s, 2H), 7.73 - 7.61 (d, J = 8.2 Hz, 2H), 7.50 - 7.42 (m, 1H), 7.43 - 7.29 (m, 2H), 3.03 - 2.28 (m, 6H), 2.13 1.90 (m, 6H), 1.03 - 0.68 (m, 12H), 0.61 - 0.20 (m, 30H) ppm. 2.2.6 Compound 1 and 2 A mixture of 8 or 9, dipyrenylcarbazole, 1 mol% CuI, 10 mol% diamine ligand and K3PO4 in dioxane ( 1 M) at 110°C for 24 h. The resulting light yellowish green mixture was allowed to cool to room temperature and extracted with CH2Cl2 (3 × 50 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated under reduce pressure. The crude product was purified by alumina chromatography using hexane:CH2Cl2 (3:1): (43% yield 1). 1H NMR (400 MHz, CDCl3) δ 8.75 8.65 (m, 2H), 8.54 - 8.49 (s, 2H), 8.38 – 8.35 (d, 2H), 8.27 (s, 2H), 8.23 – 8.02 (m, 14H), 7.90 – 7.87 (m, 2H), 7.83-7.79 (m, 4H),7.79 – 7.71 (s, 1H), 7.65 – 7.40 (m, 6H), 3.35 – 2.85 (m, 6H), 2.32 2.05 (m, 6H), 1.15 - 0.82 (m, 24H), 0.75 - 0.55 (m, 18H) ppm. 13C NMR (101 MHz, CDCl3) δ 156.4, 156.3, 156.1, 155.9, 153.8, 153.7, 146.1, 145.9, 145.8, 145.7, 145.4, 145.3, 144.9 , 144.7, 140.9, 140.4, 140.2, 140.1, 140.1, 139.9, 139.8 , 139.7, 139.7, 139.6, 139.1, 139.0, 138.8, 138.5, 138.1 , 138.0, 137.8, 136.2, 136.0, 135.6, 135.4, 133.6,131.8, 131.2, 130.6, 129.3, 129.1, 128.3, 127.6, 127.4, 126.9 , 126.8, 126.3, 126.1, 125.8, 125.2, 125.2, 124.9, 123.9, 122.8, 122.5, 120.9, 110.1, 92.9, 92.7, 92.6, 56.4, 55.8, 36.9, 36.6 , Pure and Applied Chemistry International Conference 2015 (PACCON2015) 254 29.9, 26.9, 26.7, 23.1, 22.8, 14.0 ppm. MALDITOF MS: C95H89N found 1244.909 ([M]+ calcd: 1244.702) (38% yield 2). 1H NMR (400 MHz, CDCl3) δ 8.79 8.68 (m, 1H), 8.53 - 8.84 (s, 4H), 8.39 – 8.36 (d, 4H), 8.27-8.25 (s, 4H), 8.23 – 8.02 (m, 28H), 7.97 – 7.91 (s, 4H), 7.88-7.79 (m, 8H),7.79 – 7.71 (s, 2H), 7.59 – 7.41 (m, 3H), 3.25 – 3.06 (m, 6H), 2.43 – 2.28 (m, 6H), 0.87 – 0.84 (m, 12H), 0.69 – 0.55 (m, 24H) ppm. 13C NMR (101 MHz, CDCl3) δ 156.6, 156.5, 156.4, 156.3, 154.1, 154.0, 146.3, 146.2, 146.19, 146.1, 145.9, 145.8 , 141.3, 140.6, 140.5, 140.3, 140.2, 140.1, 140.0, 139.9, 139.8, 139.7, 139.5, 138.9, 138.6 , 138.6, 138.4, 138.4, 136.6, 134.0, 132.1, 131.6, 131.3, 130.93, 129.6, 129.4, 129.2, 128.7, 127.9, 127.8, solution in 0.01 M H2SO4 (ΦF = 0.54) as a standard are 0.72 and 0.59, respectively I I a N H b N H 98% 67% N H 4 3 5 Key: (a) KI, KIO3,AcOH, refluxing, 20 min; (b) Pyrene-1-boronic acid, [Pd(PPh3)4], 2M K2CO3, refluxing THF, 24 h. Scheme 1. Synthesis of 3,6-di(pyren-1-yl)-9Hcarbazole R R CH2CH2COOH 127.39, 127.33, 126.9, 126.8, 126.6, 126.5, 126.5, 126.1, 125.6, 125.5, 125.2, 124.3, 123.0, 121.4 , 121.3, 110.5, 56.6, 56.5, 56.3, 39.3, 38.1, 37.8, 37.6, 37.3, 37.2, 33.3, 33.2, 32.7, 32.4, 30.9, 30.5, 29.8, 27.6, 27.4, 27.1, 26.9, 24.9, 23.9, 23.2, 14.6, 14.5, 14.4, 11.44, 11.4 ppm. MALDITOF MS: C139H112N2 found 1809.868 ([M]+ calcd: 1809.885) a b 49% 75% R R R 6 7 c 68% d R R R R R R R 3. Results and Discussion 3.2 Photophysical properties of compounds 1 and 2 The normalized absorption and emission spectra of 1 and 2 in CHCl3 are illustrated in Figure 1 and the pertinent data are collected in Table 1. The absorption band of the two-branched dipyrenylcarbazole truxene (2) appeared at a longer wavelength compared to the one-branched analog (1). This red-shift is attributed to the extension of conjugation upon the increment of the dipyrenylcarbazole unit. Compound 1 and 2 exhibited a maximum emission wavelength at 421 and 423 nm, respectively. Photoluminescence quantum yields yields of compounds 1 and 2 determined in CHCl3 solution (A<0.1) at room temperature using quinine sulfate 64% I I R R R R 8 3.1 Synthesis of 1 and 2 We began with a regioselective diiodination of carbazole using KI/KIO3 in refluxing acetic acid, followed by a Suzuki coupling with the commercially available pyrene-1-boronic acid to afford compound 5 in 66% yield (Scheme 1). Hereupon truxene was prepared from 3phenylpropionic acid according to the reported procedure then the methylene units were alkylated by excess amount of n-butylbromide to afford 7 in 75%. This hexaalkylated compound exhibit greater solubility in organic solvents, and also prevent the aggregation by pi-stacking leading to low quantum efficiency. Iodination of 7 with 1 eq. of HIO3 and I2 produced the core compound 8 and 2 eq. of the same reagents produced the core compound 9 in 68% and 64% yield, respectively (Scheme 2). The target compounds 1 and 2 were then accomplished by the Cu-catalyzed C-N coupling of 5 with 8 and 9 in 43% and 38% yield, respectively. R e 9 N N R R R R R R I f 38% 43% R R R R R R R N Compound 1 : R = nC4H9 Compound 2 : R = nC4H9 Key: (a) PPA, 160°C, 3 h; (b) n-BuBr, NaH, DMF, RT, 24 h; (c)(d) HIO3 and I2, CH3COOH:H2SO4:H2O:CCl4 , 80°C, 4 h; (e)(f) dipyrenylcarbazole, 1 mol% CuI, 10 mol% diamine ligand, K3PO4, dioxane ( 1 M), 110°C for 24 h. Scheme 2. Synthesis of compounds 1 and 2 Table 1: Photophysical properties of compounds 1-2 Cmpd 1 2 [a] [b] ΦF [d] Absorption Emission λmax [nm] (log ε) Solution [a] λmax [nm] 243 (4.39), 282 (4.23), 319 (4.23), 348 (4.14) 246 (4.76), 280 (4.66), 349 (4.55) 421 [b] 0.72 423 [c] 0.59 Measured in a dilute CHCl3 solution. Excited at 348 nm. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 255 [c] [d] Excited at 349 nm. Quinine sulfate in 0.01 M H2SO4 as a standard (ΦF = 0.54) 4. Conclusions We have successfully synthesized and characterized one- and two-branched dipyrenylcarbazole substituted truxene derivatives. These compounds exhibited similar absorption maxima around 348 - 349 nm and emission maxima around 421 - 423 nm in CHCl3 solution. High photoluminescence quantum yields suggested that they could serve as good emissive materials in OLEDs application. [11] Earmrattana, N., Sukwattanasinitt, M. and Rashatasakhon, P., 2012, Dyes Pigments., 93, 14281433. [12] Gomez-Lor, B., Defrutos, O., Ceballos, P.A., Granier, T. and Echavarren, A.M., 2001, Eur. J. Org. Chem., 2107-2114. [13] a) Klapars, A., Huang, X. and Buchwald, S.L., 2002, J. Am. Chem. Soc., 124, 7421 – 7428; b) Klapars, A., Antilla, C.J., Huang, X. and Buchwald, S.L., 2001, J. Am. Chem. Soc., 123, 7727 – 7729. Figure 1. Normalized absorption and emission spectra of 1 and 2 in CHCl3 solution. Acknowledgements project is supported by the This Ratchadapiseksomphot Endowment Fund of Chulalongkorn University (RES560530125-AM). References [1] Hide, F., Diaz-Garcia, M.A., Schartz, B.J. and Heeger, A.J., 1997, Acc. Chem. Res., 30, 430–436. [2] Tang, C.W. and VanSlyke, S.A., 1987, Appl. Phys. Lett., 51, 913–915. [3] Tang, C., Liu, F., Xia, Y.-J., Lin, j., Xie, L.-H., Zhong, G.-Y., Fan, Q.-L. and Huang, W., 2006, Org. Elec., 7, 155-162. [4] Li, J., Liu, D., Li, Y., Lee, C.-S., Kwong, H.-L. and Lee, S., 2005, Chem. Mater., 17, 1208-1212. [5] Promarak, V., Kochapradist, P., Prachumrak, N., Tarsang, R., Keawin, T., Jungsuttiwong, S. and Sudyoadsuk, T., 2013, Tetrahedron Lett., 54, 35833687. [6] Thomas, K.R.J., Lin, J.T., Tao, Y.T. and Ko, C.W., 2001, J. Am. Chem. Soc., 123, 9404. [7] Yang, Z., Xu, B., He, J., Xue, L., Guo, Q., Xia, H. and Tian, W., 2009, Org. Elec., 10, 954–959. [8] Kumchoo, T., Promarak, V., Sudyoadsuk, T., Sukwattanasinitt, M. and Rashatasakhon, P., 2010, Chem.-Asian J., 5, 2162-2167. [9] Huang, J., Xu, B., Su, J.-H., Chen, C.H. and Tian, H., 2010, Tetrahedron., 66, 7577-7582. [10]Raksasorn, D., 2012, Derivatives of carbazole and truxene for organic light-emitting diodes, Master's Thesis, Chulalongkorn University. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 256 CYCLIC ALKYL SULFIDE DIETHER DERIVATIVE AS AN ELECTRON DONOR FOR ZIEGLER-NATTA CATALYST IN OLEFIN POLYMERIZATION Kananat Naksomboon1, Krittaphat Wongma1, Phairat Phiriyawirut2 and Tienthong Thongpanchang1* 1 Department of Chemistry, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand 2 The Siam Cement Pcl., Bangsue, Bangkok, 10800, Thailand * E-mail: tienthong.tho@mahidol.ac.th, Tel. +66 2201 5130, Fax. +66 2201 5139 Abstract: Electron donor or Lewis base is one of the key components in propylene polymerization using ZieglerNatta catalyst. In this research, a novel electron donor based on 1,3-diether conjugated with cyclic alkyl sulfide framework was designed and synthesized and it was introduced to the polymerization reaction of propylene in a lab scale. The resulting PPs were characterized and compared to the systems without electron donor and with commercial electron donor, 2,2-diisobutyl-1,3-dimethoxypropane (DIBDMP). By using DIBDMP as a benchmark, isotacticity of PP from sulfur donor 1 is similar, with 92% of mmmm characterized by 13C NMR analysis and 91% of I.I. determined from the weight of insoluble polymers. , , and Polymer melting point (Tm) is 158 °C and MWD are 2.89 × 104, 4.63 × 105, and 16.02, respectively. These numbers are inferior as compared to the commercial donor but similar to the PP from the system without donor. The activity of the catalyst donor 1 is 1943 gPP/gTih. 1. Introduction Polypropylene (PP) is one of the most important polyolefins because of its diverse properties such as transparency, density, and tensile strength[1-2].Such polymer is generally prepared on industrial scale using Ziegler-Natta (ZN) catalyst on MgCl2 support and more than 50 million tons of isotactic polypropylene (iPP), which is semi-crystalline polymer, are produced annually from this catalyst system[3]. The stereochemistry of the resulting polymer is one of the most important factors that determine the processability, mechanical properties and chemical resistance[4]. To control the tacticity of PP, electron donors or Lewis bases are often added to regulate the polymerization process[5]. Various types of donors such as monoesters[6], phthalate esters[7], alkoxysi-lanes[3], and 1,3-diethers[7-8] have been reported and studied for propylene polymerization (Figure 1). Many models were proposed to account for the influence of electron donor on PP properties but the widely accepted one is the three-site model described by Busico et al. in which electron donors could coordinate to metal atoms (Ti or Mg) neighboring to Ti active site to control the alignment of the incoming propylene[9]. The Ti active site can be obtained by reducing pre-catalyst (Ti4+) to by co-catalyst (e.g. active species (Ti3+) triethylaluminum (AlEt3))[10]. O O OR R' R' OR OR R' R' RO OR Si RO OR O Monoesters Phthalate esters Alkoxysilanes 1,3-Diethers R, R' = alkyl, aryl Figure 1. General structures of monoesters, phthalate esters, alkoxysilanes, and 1,3-diethers. Among these electron donors, the structures with 1,3diether scaffold displayed intriguing properties since they can bind strongly to the surface of the solid support in the presence of co-catalyst, rendering a stable catalyst structure[11]. Moreover, this type of donors enhances the catalyst activity, isospecificity of polymer, and narrower molecular weight distribution (MWD) than others[12]. Due to the ability of electron donors to tune the polymer properties through the control of polymerization process, this research focuses on the synthesis of novel electron donor 1 and its role in polymerization. This electron donor was designed based on 1,3-diether which is expected to strongly adhere to the catalyst surface. The incorporation of sulfur atom in the structure is to tune the stereospecificity and catalytic activity via the donation of electron from sulfur to the solid support either through bond or space. S S OMe MeO 1 Figure 2. Structure of the designed electron donor 1 grafted with sulfur. 2. Materials and Methods 2.1 Materials TiCl4 on MgCl2 support, AlEt3, propylene gas, and 2,2-diisobutyl-1,3-dimethoxypropane(DIBDMP) were provided by The Siam Cement Group (SCG). Hexanes, methanol (MeOH), dichloromethane (DCM), ethyl acetate (EtOAc), and n-heptane were from RCI Labscan. 1,2-Ethanedithiol was from Fluka. Sodium methoxide (NaOMe), oxone, 2-iodobenzoic acid, boron Pure and Applied Chemistry International Conference 2015 (PACCON2015) 257 trifluoride diethyl etherate (BF3.Et2O), epichlorohydrin were from Aldrich. and dried over anhydrous Na2SO4, filtered, and evaporated to dryness. Column chromatography on silica gel using EtOAc and hexanes (EtOAc : hexanes, 1 : 9 v/v) as an eluent gave sulfur electron donor model 1 (0.43 g, 65% yield): 1H NMR (CDCl3) δ 3.25 (s, 4H), 3.42 (s, 6H), 3.66 (s, 4H); 13C NMR (CDCl3) δ 38.7, 59.4, 68.4, 76.9; ESI cald for C7H14NaO2S2+ [M+Na]+ = 217.0333. Found [M+Na]+ = 217.0349. 2.2 Synthesis of sulfur electron donor 1 2.2.1 Synthesis of alcohol 3 Epichlorohydrin 2 (11.50 g) was added dropwise to a solution of NaOMe (16.80 g) in anhydrous MeOH (130 mL). After 2 h, the reaction was evaporated to dryness and the mixture was extracted with DCM and washed with water (3x50 mL). The organic layer was dried over anhydrous sodium sulfate (Na2SO4), filtered, and evaporated to dryness[13]. The crude product was purified by distillation to afford a clear solution of 3 (10.40 g, 70% yield): 1H NMR (CDCl3) δ 3.39 (s, 6H), 3.41-3.48 (m, 4H), 3.93-3.99 (m, 1H); 13C NMR (CDCl3) δ 58.6, 68.6, 73.6; ESI cald for C5H12NaO3+ [M+Na]+ = 143.0684. Found [M+Na]+ = 143.0699. O NaOMe, MeOH Cl reflux, 2 h, 70% 2 3 2.2.2 Synthesis of ketone 4 To the solution of alcohol 3 (0.51 g) in EtOAc (30.0 mL) was added 2-iodoxybenzoic acid (IBX)[14] (3.59 g). The reaction was heated to reflux for 5 h and then cooled to room temperature, filtered and washed with EtOAc[15]. The filtrate was evaporated to dryness to obtain the product in 82% yield (0.41 g): 1H NMR (CDCl3) δ 3.42 (s, 6H), 4.18 (s, 4H); 13C NMR (CDCl3) δ 59.5, 76.2, 205.7; ESI cald for C5H10NaO3+ [M+Na]+ = 141.0528. Found [M+Na]+ = 141.0549. OH OMe IBX, EtOAc OMe reflux 5 h, 82% 4 3 OMe MeO 1 2.4 Characterization of PP The isotacticity of polymers was characterized by high temperature 13C nuclear magnetic resonance spectroscopy (13C NMR) and n-heptane extraction technique. The polymer molecular weight was determined by gel permeation chromatography (GPC). Melting temperature (Tm) and crystallization temperature (Tc) were obtained from differential scanning calorimetry (DSC). O MeO 0 C, 2 h, 65% 2.3 Polymerization of PP To the solution of hexanes (100 mL) was added AlEt3 1.25 mL (1.0 M in heptane) under nitrogen atmosphere and then the reaction was saturated with propylene gas at 1 atm. The electron donor (0.1 M, 1.25 mL) was introduced into the reaction followed by the addition of Ti catalyst slurry (Ti : donor : Al = 1 : 3 : 30). The reaction was stirred under propylene blanket at 30 oC. After 30 min, the reaction was quenched with 25% HCl in MeOH solution to precipitate PP. The resulting PP was washed with water (3x500 mL) and dried under reduced pressure at 60 °C for 6 h or until the constant weight was obtained. Scheme 1. The synthesis of alcohol 3. MeO S S o Scheme 3. The synthesis of sulfur electron donor 1. OMe MeO 1,2-ethanedithiol BF3.Et2O, DCM 4 OH O OMe MeO Scheme 2. The synthesis of ketone 4. 3. Results and Discussion 2.2.3 Synthesis of thioacetal derivative 1 BF3·Et2O (0.43 mL) was added to the mixture of ketone 4 (0.40 g) and 1,2-ethanedithiol (0.30 mL) in DCM (20 mL) at 0 °C. The reaction was stirred at the same temperature for 2 h and then quenched by 5% NaOH (10 mL), and extracted with DCM (3x20 mL). The combined organic layers were washed with water, The sulfur electron donor 1 could be synthesized from commercially available starting materials in 3 steps through Williamson ether synthesis, oxidation of alcohol to provide ketone, and thioacetal formation giving 37% yield overall. The polymerization of propylene was performed in hexanes with electron donor 1 and with commercialTable 1: Effects of electron donors on the catalytic activities and polymer properties. Donor PP weight (g) Activity (gPP/gTih) %mmmma %I.I.b - 2.56 2,528 85 DIBDMP 2.57 2,519 93 MWDc Tmd Tcd 2.78 16.37 159 114 5.27 12.22 160 115 (x105)c (x104)c 85 4.55 95 6.46 1.96 1,943 92 91 4.63 2.89 16.02 158 115 1 %mmmm was characterized by 13C NMR. b %I.I. was determined from Soxhlet extraction of 0.5 g of product using n-heptane as solvent under nitrogen atmosphere for 6h. c , , and MWD were characterized by GPC. d Tm and Tc were characterized by DSC. a Pure and Applied Chemistry International Conference 2015 (PACCON2015) 258 ized electron donor DIBDMP (Figure 3) and in the absence of electron donor for the purpose of comparison. The catalyst activity and properties of the resulting PPs are summarized in Table 1. MeO OMe DIBDMP Figure 3. The structure of commercially used donor in industry (DIBDMP). The effects of electron donors on the catalytic activities were investigated by the determination of the weight of PP. The polymerizations with commercial electron donor, i.e. DIBDMP, and without gave similar PP weight implying that the catalytic activities were not affected by the addition of electron donor. In contrast, the use of sulfur donor 1 as an electron donor decreased both the PP weight and activity to 1.96 g and 1,943 gPP/gTih, respectively. This is presumably due to the poisoning of sulfur atom of electron donor to the Ti catalyst[16]. a) 1 2 CH2 CH CH3 3 in comparison to other stereoisomers[16]. As shown in Figure 4a, the spectrum features 3 main peaks of PP carbons labeled with numbers corresponding to the labeled carbon in the monomeric propylene structure. To calculate %mmmm, the integrated peak area of methyl carbon or C3 labeled as mmmm in the expanded NMR spectrum (Figure 4b) is determined as percentage in comparison to the integrated peak areas of other stereoisomers[17]. Another parameter to indicate isotacticity of PP is %isotacticity index (%I.I.), this data can be obtained by determining a fraction of insoluble polymer in n-heptane which is isotactic PP to the overall polymer weight. %mmmm increases from 85, 92 to 93 and %I.I. increases from 85, 91 to 95 for the reactions with no donor, sulfur donor and commercial donor, respectively. This result indicates that the polymers obtained from the reactions with electron donors are isotactic[18]. Although the reaction with sulfur electron donor 1 displayed low catalytic activity, a comparable stereoselectivity was still obtained. The number average molecular weight ( ) and weight average molecular weight ( ) were determined from GPC. The polymer obtained from the reaction without donor and with sulfur electron donor 1 and in comparison to commercial yielded lower electron donor. In addition, PP synthesized from the reaction with DIBDMP displayed the narrowest distribution as compared to the reaction with sulfur donor and no donor. The crystallinity of the PPs was evaluated from Tm and Tc obtained from DSC analyses. Tm and Tc of all PPs from the reaction with and without donors are quite similar indicating that the obtained PPs are of comparable crystallinity. 4. Conclusions The sulfur electron donor 1 was successfully synthesized in 3 steps in satisfactory yield. This novel electron donor was introduced to the propylene polymerization reaction and the obtained polymer , and showed high %mmmm and %I.I. while the are not better than that of the reaction with DIBDMP. b) Acknowledgements Figure 4. 13C NMR spectrum of PP obtained using sulfur derivative 1 as electron donor (a) PP 13C NMR (b) the expanding spectrum of methyl carbon (C3). Financial support from Development and Promotion of Science and Technology talents project (DPST) and facilities support from the Center of Excellence for Innovation in Chemistry (PERCH–CIC) are gratefully acknowledged. Authors also thank SCG for materials and PP characterization and Dr. Tossapol Khamnaen for valuable suggestion. The other important characteristic of PP is tacticity since it is related to the physical properties of the polymer. The polymer tacticity was identified from 13C NMR by determining the %pentad tacticity (%mmmm) Pure and Applied Chemistry International Conference 2015 (PACCON2015) 259 References [1] Zhang, S. and Horrocks, A.R., 2003, Prog. Polym. Sci., 28, 1517-1538. [2] Marques, M.d.F.V., Cardoso, R.d.S. and da Silva, M.G., 2010, Appl. Catal. A, 374, 65-70. [3] Shen, X.-R., Fu, Z.-S., Hu, J., Wang, Q. and Fan, Z.-Q., 2013, J. Phys. Chem. C, 117, 15174-15182. [4] Zhao, S., Chen, F., Huang, Y., Dong, J.-Y. and Han, C.C., 2014, Polymer, 55, 4125-4135. [5] Lu, L., Niu, H. and Dong, J.-Y., 2012, J. Appl. Polym. Sci., 124, 1265-1270. [6] Liu, B., Cheng, R., Liu, Z., Qiu, P., Zhang, S., Taniike,T., Terano, M., Tashino, K. and Fujita, T., 2007, Macromol. Symp., 260, 42-48. [7] Sacchi, M. C., Forlini, F., Tritto, I., Locatelli, P., Morini, G., Noristi, L. and Albizzati, E., 1996, Macromolecules, 29, 3341-3345. [8] Cui, N., Ke, Y., Li, H., Zhang, Z., Guo, C., Lv, Z. and Hu, Y., 2006, J. Appl. Polym. Sci., 99, 1399-1404. [9] Liu, B., Nitta, T., Nakatani, H. and Terano, M., 2004, Macromol. Symp., 213, 7-18. [10] Arlman, E. J. and Cossee, P., 1964, J. Catal., 3, 99-104. [11] Xu, D., Liu, Z., Zhao, J., Han, S. and Hu, Y., 2000, Macromol. Rapid Commun., 21, 1046-1049. [12] Malpass, D.B. and Band, E., 2012, Introduction to Industrial Polypropylene: Properties, Catalysts Processes, Wiley. [13] Bebernitz, G.R., Dain, J.G., Deems, R.O., Otero, D.A., Simpson, W.R. and Strohschein, R.J., 2000, J. Med. Chem., 44, 512-523. [14] Frigerio, M., Santagostino, M. and Sputore, S., 1999, J. Org. Chem., 64, 4537-4538. [15] More, J.D. and Finney, N.S., 2002, Org. Lett., 4, 30013003. [16] Dunleavy, J.K., 2006, Platinum Metals Rev., 50, 110. [17] Asakura, T., Demura, M. and Nishiyama, Y., 1991, Macromolecules, 24, 2334-2340. [18] Brintzinger, H.H., Fischer, D., Mülhaupt, R., Rieger, B. and Waymouth, R.M., 1995, Angew. Chem. Int. Ed. Engl., 34, 1143-1170. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 260 ΑLPHA-BROMINATION OF KETONE USING HEXABROMOACETONE Tipakorn Sangrawee1, Warinthorn Chavasiri2,* 1 Program in Petrochemistry and Polymer Science, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand 2 Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand * Email: warinthorn.c@chula.ac.th Abstract: An efficient method for the preparation of αbromoketone has been disclosed. Propiophenone, a model substrate was treated with hexabromoacetone (HBA) with the ratio of substrate to HBA as (1:1) in THF at RT for 5 min under UV (254 nm) irradiation. 2Bromopropio- phenone was obtained as a sole product in high yield. Various parameters including reaction time, solvent and ratio of substrate to HBA were scrutinized. 1. Introduction α-Bromination of carbonyl compounds is one of important transformations in organic synthesis as αbrominated products are useful synthetic intermediates.1 Previously, α-brominated carbonyl compounds could be prepared by using Br2 in protic solvent in the presence of Lewis acid,2 Nbromosuccinimide (NBS)3 and cupric bromide.4 Recently, various methods have been reported using NBS-NH4OAc,5 NBS-photochemical,6 NBS-silica supported sodium hydrogen sulfate,8 and NBSPTSA,11 However, the disadvantages of those procedures are in some cases the substrates or products being intolerance to acid, requiring high temperature and forming undesired by-product. In 1969, HBA was first synthesized; nevertheless without utilizing as a reagent in organic chemistry.12 Until 2008, our research group explored the utilization of this reagent coupled with PPh3 as a new and efficient brominating agent for alcohol under mild conditions with short reaction time.13 In 2009, other researchers reported the use of this reagent as tribromoacetylating agent of alcohols and amines and as mediator in the conversion of carboxylic acid into amides14 with moderate to good yields. In 2011, another group addressed the preparation of benzyl bromides from alcohols with high conversion rates and short reaction time.15 Up to date, HBA has not been employed as a brominating agent for α-bromination of ketone. Herein, we report a novel, mild and high yielding synthetic method for α-bromination of ketone by using propiophenone as a model substrate. 2. Materials and Methods Instrumental: Thin layer chromatography (TLC) was performed on aluminum sheets pre-coated with silica gel (Merck Kieselgel 60 PF254). Column chromatography was carried out on silica gel (Merck Kieselgel 60, 70-230 mesh). The 1H-NMR spectra were performed in CDCl3 with tetramethylsilane (TMS) as an internal reference on a Varian nuclear magnetic resonance spectrometer, model Mercury plus 400 NMR spectrometer which operated at 399.84 MHz for 1H. Chemicals: All solvents were purified by standard methods before use unless those were reagent grades. The reagents for synthesis were purchased from Fluka or Sigma-Aldrich and used without further purification. General procedure: In a quartz cell (cylinder tube 2.5 x 13.5 cm), propiophenone 17 µL (0.125 mmol) was mixed with HBA 67 mg (0.125 mmol) in THF (1 mL). The reaction mixture was stirred at RT under the irradiation of UV light (254 nm, 6W) for 5 min (Scheme 1). The reaction was monitored by TLC and at appropriate time was quenched by adding NaHCO3 (1 mL) and extracted twice with Et2O (5 mL). The product yield was analyzed by GC using naphthalene as an internal standard. O O Br3C CBr3 0.125 mmol O THF, UV (254 nm), 5 min Br 0.125 mmol Scheme 1. α-Bromination of propiophenone using HBA 3. Results and Discussion Table 1 reveals the effect of types of brominating agent on the conversion of propiophenone into 2bromopropiophenone. The reaction with HBA provided a high yield of the desired product (entry 1) while other common brominating agents (entries 2-6) gave low yields of the target molecule. Table 1: Effect of types of brominating agent Entry Brominating % Recovery of % agent propiophenone Yield 1 HBA 38 72 2 NBS 94 0.1 3 CBr4 90 17 4 Br3CCO2H 88 10 5 C2H5Br 97 0 6 BrCCl3 99 7 Reaction conditions: Propiophenone (0.125 mmol), brominating agent (0.125 mmol), Et2O (1 mL) 5 min, UV, RT Pure and Applied Chemistry International Conference 2015 (PACCON2015) 261 Effect of reaction time with UV-irradiation As presented in Table 2, the reaction in the absence of UV light (entry 1) generated the target product in poor yield. The yield of this product increased when the reaction was prolonged. The optimal reaction time was observed to be 10 min (entries 2-4). The yield decrease while the undesirable formation of α,α-dibromination product increased after prolonged irradiation to be 15 min (entry 4). This result clearly demonstrated that irradiation by UV light was important for this reaction. Table 2: Effect of reaction time with UV-irradiation Entry Reaction % Recovery of % time (min) propiophenone Yield 1 5 98 5 (without UV) 2 5 38 72 3 10 6 100 4 15 6 94 Reaction conditions: Propiophenone (0.125 mmol), HBA (0.125 mmol), Et2O (1 mL), reaction time and UV (vary), RT target product based on the amount of HBA, the formation of 2-bromopropiophenone was increased when low amount of HBA was employed. In addition, this experiment was set up to observe the efficiency of HBA. In entry 1, with the mole ratio of propiophenone to HBA 1:1, the target product could be attained in very high yield based on the mole of brominating agent used. These results displayed a good efficiency of this brominating agent. Table 5: Effect of molar ratio of propiophenone: HBA Molar ratio of propiophenone: HBA % Yield % Recovery of propiophenone Based on HBA 1 1.00: 1.00 3 97 97 2 14 81 163 2.00: 1.00 3 32 57 170 3.00: 1.00 4 36 63 377 6.00: 1.00 Reaction conditions: propiophenone:HBA (vary), THF (1 mL), 5 min, UV, RT Entry Based on propiophenone Step I (Initiation) Effect of solvent As displayed in Table 3, the reaction in Et2O generated the target product in poor yield (entry 1), while THF provided the best result (entry 6). O Br Br Br Attempting to reduce the reaction time in THF to 2 min did not give good yield of the desired product (Table 4, entry 1). Thus, the optimal reaction time should be 5 min. Table 4: Effect of reaction time with UV-irradiation Entry Reaction % Recovery of % Yield time (min) propiophenone 1 2 36 69 2 5 3 97 Reaction conditions: Propiophenone (0.125 mmol), HBA (0.125 mmol), THF (1 mL), UV, RT Effect of the molar ratio of propiophenone: HBA Table 5 reveals that when the ratio of propiophenone to HBA was increased or using less HBA, the yield of the desired product was decreased. This was clearly demonstrated that the amount of HBA was essential for this reaction. Considering the yield of Br Br Br Br Br Br O O Br H H Table 3: Effect of solvent Entry % Recovery of solvent % Yield propiophenone 1 Et2O 98 5 2 DCE 98 6 3 DCM 95 4 4 Hexane 75 11 5 CH3CN 62 38 6 THF 3 97 7 Benzene 100 5 Reaction conditions: Propiophenone (0.125 mmol), HBA (0.125 mmol), solvent (1 mL), 5 min, UV, RT O Br Br Br HBr H Step II (Propagation) O O Br Br H Br O O Br Br Br H Br Br Br O O O BrCCCBr3 H H Br H O BrCCCBr3 H Step III (Termination) O O Br Br H Br Br O Br Br H Br O Br3CCCBr2 H H Br O Br2CCCBr2 H O Br2CCCBr2 H H H Br Br2 Scheme 2. The proposed mechanistic pathway for αbromination of propiophenone using HBA 4. Br Br Br Conclusions HBA was disclosed as an efficient brominating agent for a clean and rapid α-bromination of propiophenone under the irradiation of UV light with short reaction time. This bromination reaction could proceed cleanly and rapidly to give the desired product in excellent yield under mild conditions. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 262 References [7] [1] [2] [8] [3] [4] [5] [6] Garbisch, E.W., 1965, J. Org. Chem., 30, 2109 (a) Boyd, R.E., Rasmussen, C.R. and Press, J.B., 1995, Synth. Commun., 25, 1045-1051. (b) Karimi, S. and Grohmann K, G., 1995, J. Org. Chem., 60, 554-559. (c) Curran, D.P., Bosch, E., Kaplan, J. and Comb, M.N., 1989, J. Org. Chem., 54, 1826-1831. (d) Curran, D.P. and Chang C T. 1989, J. Org. Chem., 54, 31403157. (a) Coats, S.J. and Wasserman, H.H., 1995, Tetrahedron Lett., 36, 7735-7738. (b) Shi, X. and Dai, L.J., 1993, J. Org. Chem., 58, 4596-4598. King, L.C. and Ostrum, G.K., 1964, J. Org. Chem., 29, 470-471. Lee, J.C., Bae, Y.H. and Chang, A.K., 2003, Bull. Korean Chem. Soc., 24, 407-408. Arbuj, S.S., Waghmode, S.B. and Ramaswamy, A.V., 2007, Tetrahedron Lett., 48, 1411-1415. [9] [10] [11] [12] [13] [14] [15] Das, B., Venkateswarlu, K., Mahender, G. and Mahender, I., 2005, Tetrahedron Lett., 46, 3041-3044. Guha, S.K., Wu, B., Kim, B.S., Baik, W. and Koo, S., 2006, Tetrahedron Lett., 47, 291-293. Lee, J.C. and Park, H.J., 2007, Synth. Commun., 37, 87-90. Pravst, I., Zupan, M. and Stavber, S., 2008, Tetrahedron, 64, 5191-5199. Zavozin, A.G., Kravchenko, N.E., Ignat’ev, N.V. and Zlotin, S.G., 2010, Tetrahedron Lett., 54, 545-547. Gilbert, E.E., 1969, Tetrahedron, 25, 1801-1806. Tongkate, P., Pluempanupat, W. and Chavasiri, W., 2008, Tetrahedron Lett., 49, 1146-1148. Menezes, F.G., Kolling, R., Bortoluzzi, A.J., Gallardo and H., Zucco, C., 2009, Tetrahedron Lett., 50, 25592561. Joseph, K.M. and Sanchez, I.L., 2011, Tetrahedron Lett., 52, 13-16. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 263 FRIEDEL–CRAFTS BENZYLATION OF TOLUENE USING NdCl3 IMPREGNATED ON ALUMINIUM PILLARED MONTMORILLONITE Natthida Maneechandra1, Warinthorn Chavasiri2* 1 Program in Petrochemistry and Polymer Science, 2Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand * E-mail: warinthorn.c@chula.ac.th, Tel. 02-2187625, Fax 02-2187598 Abstract: 2%NdCl3 impregnated on aluminium pillared montmorillonite (2%NdCl3/Al-PLM) was prepared as a catalyst for Friedel–Crafts benzylation of toluene with benzyl alcohol. The appropriate amount of 2%NdCl3/AlPLM was investigated. Its basal spacing was characterized by XRD technique. The reaction was performed using 0.8%mol of 2%NdCl3/Al-PLM at 170°C for 1 h with 8:1 molar ratios of toluene and benzyl alcohol. This reaction yielded quantitative yield of benzyl toluene with a ratio ortho:para of 1.52. In addition, this clay catalyst was easy to separate and could be reused for at least 5 times without losing catalytic activity. OH 0.8% mol of 2% NdCl /Al-PLM 3 + toluene benzyl alcohol + + sealed tube, 170oC, 1 h o-benzyl toluene p-benzyl toluene O benzyl ether Keywords NdCl3; Aluminium Pillared Montmorillonite; Clay; Friedel-Crafts Benzylation 1. Introduction Lewis acid clay has been used as a catalyst in various reactions for years. It has also widely become interest due to its advantages such as the environmental friendliness and the applicability in the desired catalytic condition.1 There are several methods to develop the clay catalyst such as cation-exchange, pillaring and impregnation. However, the activity of these clay catalysts depends on the nature of the metal cation in acid treated clays.2 Herein, we observed high activity of NdCl3 impregnated on aluminium pillared montmorillonite (NdCl3/Al-PLM), in benzylation of toluene. Friedel-Crafts benzylation of aromatics is an important reaction for generating a wide variety of diphenylmethane derivatives which are key synthetic intermediates in pharmaceutical and fine chemical industries. Conventionally, these benzylations are carried out industrially by dangerous homogeneous acid catalysts such as AlCl3, BF3, H2SO4, HF, HNO3, etc. These catalysts generate high volume of waste materials. Moreover, they also pose several problems such as corrosion, toxicity, difficulty in separation, disposal, and reusability of the catalysts.3-7 The present work aims to develop a highly efficient and reusable catalyst by using 2% NdCl3/Al-PLM for the selective Friedel-Crafts benzylation between toluene and benzyl alcohol to afford diphenylmethane. Herein, we reported simple preparation of this catalyst, the appropriate amount of catalyst, temperature, time, mole ratio, and reusability cycle number. In addition, a comparison of the benzylating agents was also described. 2. Materials and Methods 2.1 Materials and reagents Bentonite clay (Cernic International Co. Ltd.) with the typical chemical analysis of (%wt): SiO2 63.60, Al2O3 17.60, Fe2O3 3.10, CaO 3.00, Na2O 3.40, K2O 0.50, was purified by fractionated centrifugation in order to discard quartz and impurities to get montmorillonite. Then, montmorillonite from the previous step was suspended in 5 M NaOH with the ratio of clay to NaOH solution as 1 g : 50 mL for 24 h at room temperature and repeated for three times to get Na-montmorillonite. For the preparation of catalysts, AlCl3⋅6H2O (Sigma Aldrich), NaOH (Merck) and NdCl3⋅6H2O (Sigma Aldrich) were of laboratory grade. De-ionized water was used throughout the study. The prepared catalysts were characterized by a X-ray powder Rigaku, Dmax 2200/utima+ diffractometer with a monochromater and Cu K ∝ radiation. The 2θ was ranged from 2 to 30° with the scan speed of 3°/min and the scan step of 0.02°.8 For catalytic study, toluene (Merck), benzyl alcohol (Carlo ERBA), benzyl ether (Fluka), benzyl chloride (Merck) were purchased as laboratory reagent grade. Products were monitored by Shimadzu GC (FID, SGE BP1 column). 2.2 Preparation and characterization of 2%NdCl3/AlPLM Al-PLM was synthesized by the intercalation aluminium pillaring precursors, followed by calcination at high temperature. Na-clays were dispersed in deionized water (10%wt) by continuous stirring for 1 day at room temperature. Al-pillaring agent was prepared by hydrolysis of NaOH and AlCl3⋅6H2O with a mole ratio of OH/Al 1.9. Then, the mixture was added slowly to the clay suspension for 24 h at room temperature. The products were collected by centrifugation and washed with deionized water until chloride ions were eliminated, checked the disappearance of chloride ion by AgSO4. The intercalated product (Al-PLM) was dried at 100°C for 24 h and calcined at 500°C at the rate of 5°C/min for 1 Pure and Applied Chemistry International Conference 2015 (PACCON2015) 264 h. Next, the Al-PLM was impregnated by using a solution of 2%NdCl3 in amount of minimized EtOH. The slurry mixture was dried at 60°C and calcined at 450°C at the rate of 5°C/min for 4 h to obtain 2%NdCl3/Al-PLM.8 2.3 General benzylation procedure In a sealed tube with a magnetic stirring bar, 2% NdCl3/Al-PLM, benzylating agent 0.5 mmol and toluene 8 mmol were mixed and heated. The reaction mixture was filtered and washed with acetone. The benzylation products were quantified by GC analysis. Then, catalyst was activated by calcination at 450 °C at the rate 5°C/min for 4 h. 3. Results and Discussion 3.1 Characterization of catalysts Figure 1 represents the XRD patterns of montmorillonite (a), Na-montmorillonite (b), Al-PLM (c), and 2%NdCl3/Al-PLM (d) of 001 peak at 2θ. Namontmorillonite (b) shows peak shifted to lower 2θ comparing with montmorillonite (a), while d001 basal spacing of Na-montmorillonite (b) became increasing of 11.4 Å to 12.5 Å. The different clearance space of clay layers was resulted by solvating of Na ions dispersion for balancing charge. The broad peak of AlPLM (c) was shown 13.2 Å which was shifted to the larger basal spacing comparing with the d001 peak of Na-montmorillonite (b). The resulting of larger basal spacing of Al-PLM with Na ion between the layers of montmorillonite could be exchanged by aluminium polyoxocations. The OH/Al was intercalated into the clay layer and converted to Al-pillared montmorillonite during calcination process. The d001 basal spacing of NdCl3/Al-PLM was 15.8 Å which larger than that of Al-PLM. The d001 peak was broader because of the re-calcination process which caused the pillared stucture to collapse slightly and also the impregnation of NdCl3 further increases distance between the interlayer spacing of clay. Figure 1. X-ray diffraction pattern of: (a) montmorillonite; (b) Na-montmorillonite; (c) Al-PLM; (d) 2%NdCl3/Al-PLM 3.2 Catalytic activity of 2%NdCl3/Al-PLM on FriedelCrafts benzylation of toluene The Friedel-Crafts benzylation of toluene with benzyl alcohol mainly produced benzyl toluene and benzyl ether in different pathways. The benzyl ether was formed by self-condensation of benzyl alcohols. On the other hand, benzyl toluenes were produced between toluene and benzyl alcohol by using 2% NdCl3/Al-PLM which played the important role as Lewis acid in this reaction. The effect of catalyst amount, 2%NdCl3/Al-PLM in Friedel-Craft benzylation of toluene and benzyl alcohol was investigated as shown in Table 1. The products were not observed in the absence of 2%NdCl3/Al-PLM (entry 1). However, 0.4% mol (10 mg) of 2%NdCl3/Al-PLM yielded benzyl toluene and benzyl ether (entry 2). It could be rationale that the acidity of 0.4% mol of this catalyst was too low for the production of benzyl toluene. Hence, benzyl ether was formed by self-condensation from the remaining benzyl alcohols. On the other hand, increasing amount of 2%NdCl3/Al-PLM to 0.8%mol (20 mg) and 2%mol (50 mg) affected 100% conversion of benzyl alcohol and none of benzyl ether was detected (entries 3-4). Mass balance (MB) more than 95% indicated that there was no undesired products occurred in the reaction. 3.3 The effect of reaction temperature From Table 2, the reaction temperature of 170°C gave 100% conversion of benzyl alcohol. It means that monobenzylation of toluene underwent efficiently (entry 4). However, at lower temperature, only trace products were occurred (entries 1-3). 3.4 The effect of reaction time The reaction time was investigated (Table 3). The yield of products was increased when the reaction time increased. For 15 min, high amount of recovered benzyl alcohol and 18% yield of products were observed (entry 1). When the reaction time was extended to 30 min, the products were formed almost 100% (entry 2). The better yields of products could be accomplished but little yield of benzyl ether was occurred. This could be described that benzyl ether which was formed from benzyl alcohol also functioned as a benzylating agent in the reaction. The benzylation of toluene with benzyl alcohol required 1 h for 100% conversion of substrate to benzyl toluene (entry 3). 3.5 The efffect of mole ratio Friedel-Crafts benzylation of toluene with benzyl alcohol was carried out by varying the mole ratio of toluene to benzyl alcohol as follows: 8:1, 16:1, 32:1 and 64:1 (entries 1-4). The conversion of benzyl alcohol was found to increase with the higher reactant mole ratio as shown in Table 4 (entries 2-4). Further increase in mole ratio had affected on the reduction of benzyl ether formation which is noticeable in the mole Pure and Applied Chemistry International Conference 2015 (PACCON2015) 265 ratio of 8:1 (entry 1). This means that there was no sufficient toluene to react with benzyl alcohol. Table 1: The effect of 2%NdCl3/Al-PLM amount on Friedel-crafts benzylation of toluene Amount of % products 2% NdCl3/ Mass % recovery % o-benzyl % p-benzyl % benzyl Al-PLM (% balance toluene toluene ether mol, mg) 0 96.9 0 0 0 96.9 0.4, 10 29.2 23.8 16.1 28.2 97.3 0.8, 20 0 59.3 39.0 0 98.3 2, 50 0 62.9 42.3 0 105.2 Reaction conditions: toluene (8 mmol), benzyl alcohol (0.5 mmol), sealed tube, 170 °C, 1 h o:p- (o-+p-)/ benzyl ether 1.48 1.52 1.49 1.41 - Table 2: The effect of temperature on Friedel-Crafts benzylation of toluene % products (o-+p-)/ Tempera Mass o% recovery benzyl % o-benzyl % p-benzyl % benzyl balance :pture (°C) ether toluene toluene ether RT (30) 92.6 0 0 0 92.6 70 92.2 0 0 0 92.2 120 92.2 1.0 0.7 1.2 94.4 1.43 1.42 170 0 59.3 39.0 0 98.3 1.52 Reaction conditions: toluene (8 mmol), benzyl alcohol (0.5 mmol), sealed tube, 1 h, 2%NdCl3/Al-PLM (0.8% mol, 20 mg) Table 3: The effect of time on Friedel-Crafts benzylation of toluene % products (o-+p-)/ Mass oTime benzyl % recovery % o-benzyl % p-benzyl % benzyl balance :p(min) ether toluene toluene ether 15 75.3 7.0 4.6 6.4 93.3 1.52 1.81 30 0 60.7 41.4 5.5 107.6 1.47 18.56 60 0 59.3 39.0 98.3 1.52 Reaction conditions: toluene (8 mmol), benzyl alcohol (0.5 mmol), sealed tube, 170 °C, 2%NdCl3/Al-PLM (0.8% mol, 20 mg) Table 4: The effect of mole ratio on Friedel-Crafts benzylation of toluene Ratio of % products (o-+p-)/ toluene: Mass o% recovery benzyl % o-benzyl % p-benzyl % benzyl benzyl balance :pether toluene toluene ether alcohol 8:1 15.8 33.5 22.5 32.7 104.5 1.49 3.54 16:1 0 59.3 39.0 98.3 1.52 32:1 0 62.8 37.1 99.9 1.69 64:1 0 67.1 43.7 110.8 1.53 Reaction conditions: toluene (8 mmol), benzyl alcohol, sealed tube, 170 °C,1 h, 2%NdCl3/Al-PLM (0.8% mol, 20 mg) Table 5: The effect of benzylating agent on Friedel-Crafts benzylation of toluene % products Benzylating Mass balance o-:p% recovery % o-benzyl % p-benzyl agent toluene toluene Benzyl alcohol 0 59.3 39.0 98.3 1.52 Benzyl chloride 0 58.8 42.2 101 1.39 Benzyl ether 104.3 0.6 0.3 105.2 1.81 Reaction conditions: toluene (8 mmol), benzylating agent (0.5 mmol), sealed tube, 170°C, 1 h, 2%NdCl3/Al-PLM (0.8% mol, 20 mg) 3.6 The effect of benzylating agent There was no recovery of benzyl alcohol and benzyl choride within 1 h (Table 5, entries 1-2). However, the trace products were observed when benzyl ether was a substrate due to the inefficient leaving group of benzyl ether (entry 3). Thus, the capability of benzylating Pure and Applied Chemistry International Conference 2015 (PACCON2015) 266 agent in this reaction is as follows: benzyl alcohol ≈ benzyl chloride > benzyl ether. Although both benzyl alcohol and benzyl chloride gave good yields in the first 1 h, benzyl chloride was 3.7 Recovery and reuse of 2%NdCl3/Al-PLM 2%NdCl3/Al-PLM could be easily recovered by filtration. After washing with acetone, drying at 80°C in oven and calcination at 450°C at the rate of 5°C/min for 4 h, the recovered 2%NdCl3/Al-PLM was used in the next run. The catalyst can be reused at least 5 times without appreciable loss of activity (entries 1, 2, 3, 6, 9-11). The results are shown in Table 6. The first and second reuses of the catalyst under the same optimized conditions showed 100% conversion (entries 1,4), whereas the efficiency for the third run was not good and showed 95% recovery of benzyl alcohol (entry 7). According to the results, the efficiency of catalysts was reduced when the reusability cycle numbers were increased. It could be described that 2% NdCl3/AlPLM could not display the same activity because of corrosive and released HCl. In addition, HCl formed in Friedel-Crafts benzylation could cause the structural damage of the clay catalyst.9 the high temperature in processing recovery, which may cause the structural deterioration and consequently lowered its activity. Hence, it was reasonable to add higher amount of catalyst to 4% mol (100 mg) of 2%NdCl3/Al-PLM in the next run (entries 9-14). On the other hand, the forth and fifth reused catalyst could give high yield of benzyl toluene (entries 10-11). The sixth and seventh reused catalyst still exhibited 100% conversion but the high amount of benzyl ether was detected as a result of insufficient Lewis acid in the catalytic process (entries 12-13). The eighth reused catalyst was inefficient for Friedel-Crafts benzylation of toluene with benzyl alcohol since there was 8.8% recovery of substrate benzyl alcohol (entry 14). This indicated that the catalyst can be reused at least five times without appreciable loss of activity. Table 6: Activity of regenerated 2%NdCl3/Al-PLM on Friedel-Crafts benzylation of toluene % products Amount of % 2% NdCl3/ % o% p% MB o-:pTimes Al-PLM recovery benzyl benzyl benzyl (% mol, mg) toluene toluene ether 1 0.8, 20 0 60.9 39.6 0 100.5 1.54 2, 50 0 67.1 44.1 0 111.1 1.52 4, 100 0 63.8 41.1 0 104.9 1.55 2 0.8, 20 0 58.1 37.8 5.9 101.8 1.54 2, 50 0 63.7 40.2 1.9 105.9 1.58 4, 100 0 64.2 42.1 0 106.3 1.53 3 0.8, 20 95.1 0.6 0.4 0.5 96.7 1.49 2, 50 5.6 42.6 27.8 29.8 105.8 1.53 4, 100 0 60.8 38.8 0 99.6 1.57 4 4, 100 0 61.8 40.5 0.9 103.2 1.53 5 4, 100 0 63.6 40.8 0.9 105.3 1.56 6 4, 100 0 56.5 36.9 6.5 99.9 1.53 7 4, 100 0 56.1 35.8 12.5 104.4 1.57 8 4, 100 8.8 43.1 27.4 20.9 100.2 1.57 Reaction conditions: toluene (8 mmol), benzyl alcohol (0.5 mmol), sealed tube, 170 °C, 1 h 4. Conclusions The aim of this research was to study the catalytic benzylation of toluene by using 2%NdCl3/Al-PLM as the catalyst. The catalyst was synthesized by intercalation of aluminium polyoxocations into clay layers and then impregnation of 2%NdCl3. The synthetic clay was characterized with XRD technique. For catalytic reaction, 2%NdCl3/Al-PLM exhibited highly reactive in the Friedel-Creafts benzylation of toluene and could be recovered at least five times by adding 4%mol of 2%NdCl3/Al-PLM in the next run. References [1] Ahmed, O.S. and Dutta, D.K., 2005, J. Mol. Cat. A., 229, 227-231. (o-+p)/ benzyl ether 16.19 53.69 1.92 2.36 113.83 110.71 14.33 7.38 3.37 [2] Shrigadi, N.B., Shinde, A.B. and Samant, S.D., 2003, Appl. Clay Sci. A., 252, 23-35. [3] Kumar, CH. R., Rao, K.T. V., Prasad, P.S. S. and Lingaiah, N., 2011, J. Mol. Cat. A., 337, 17-24. [4] Salavati-Niasari, M., Hasanalian, J. and Najafian, H., 2004, J. Mol. Cat. A., 209, 209-214. [5] Sreekumar, R.. and Pillai, C.N., 1993, Catal Lett., 19, 281-291. [6] Choudhary, V.R. and Jana, S.K., 2002, J. Mol. Cat. A., 180, 267-276. [7] Li, J., Xu, C., Lou, L.L., Zhang, C., Yu, K., Qi, B., Liu, S. and Wang, Y., 2013, Catal. Commun., 38, 59-62. [8] Keawbuarom, P. “Regioselectivity of phenol alkylation using metal impregnated aluminium pillared bentonite” Master’s Thesis, Department of Petrochemistry and Polymer Science, Faculty of Science, Chulalongkorn University, 2011. [9] Choudhary, V.R., Jha, R. and Narkhede, V.S., 2005, J. Mol. Cat. A., 239, 76-81. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 267 SYNTHESIS AND CHARACTERIZATION OF ISOINDIGO DERIVATIVES AS MOLECULAR DONORS FOR ORGANIC PHOTOVOLTAICS Patthira Sumsalee1, Visit Waewsungnoen2 and Vinich Promarak3* School of Chemistry, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand E-mail: pattypatthira@hotmail.com Abstract: Isoindigo was directly used as the building block for oligomers or polymers, and less attention was paid on the manipulation on its core structure. Still, modification on isoindigo core may have influence on its electric properties. In this work, we design new high performance isoindigo-containing donor molecules employing a novel molecular architecture with two isoindigo chromophores in the conjugated backbone. Three isoindigo derivatives containing aromatic cores were synthesized using a combination of aldol condensation, alkylation and Suzuki cross coupling reactions. The different core moieties, anthracene, benzothiadiazole and fluorene, were used in order to increase molar absorptivity of desired molecules. They were characterized by 1H NMR, 13C NMR, FT-IR and mass spectrometry. The optical properties were studied in dichloromethane solution. The desired compounds exhibited wide absorption spectra in UV-visible region (300-600 nm) with high molar extinction coefficient. The results suggest that the synthesized compounds can be used as donor molecules in organic photovoltaic devices. 1. Introduction Organic photovoltaics (OPVs) have achieved remarkable progress due to unique advantages such as low cost, light weight and applications in flexible large-area devices. Nitrogen-containing electrondeficient dyes, isoindigo, have attracted an increasing attention as building blocks for organic photovoltaic materials. High charge carrier mobility was obtained for conjugated polymers based on isoindigo derivatives. In this work, we synthesized and characterized the novel isoindigo derivatives as electron donor for organic photovoltaics. The different core moieties, anthracene, benzothiadiazole and fluorene, were used in order to increase molar absorptivity of desired molecules. 2. Materials and Methods 2.1 Materials and instruments Tetrahydrofuran (THF) was refluxed with benzophenone and Na. Reagent and chemical were purchased from chemical industry. 1H NMR and 13C NMR were record by a Bruker Advance 500 mHz. UV-Vis spectra were measured by Perkin-Elmer UV lambda 25 spectrometer. 2.2 Experimental section 2.2.1 (E)-6,6'-dibromoisoindigo (3) [1] A solution of 6-bromoisatin (0.5332 g, 2.21 mmol), 6bromooxindole (0.5031g, 2.36 mmol) and HCl 0.1 ml as catalyst were added in 15 ml of acetic acid. The reaction was refluxed for 15 h. The solution was cooled and poured into water to give brown solid as 96% yield. 2.2.2 Iisoindigo derivatives (4) (E)-6,6'-dibromoisoindigo (3) (0.8377g, 5.95mmol) and K2CO3 (0.8223 g 5.95 mmol) were dissolved in DMF 40 ml then 1-bromo-2-ethyl hexane 0.5 ml was added. The solution was heated at 100 oC for 15 h. The organic solvent was collected and dried over Na2SO4. The crude product was purified by column chromatography with hexane and dichloromethane as eluent (2:1) to give red solid as 84% yield. 1H NMR (500 mHz, CDCl3) δ = 9.03 (2H, d, J=10.0 Hz), 7.15 (2H, d, J=10.0 Hz), 6.89 (2H, s), 3.62 (4H, m), 1.82 (2H, s), 1.29 (16H, m) and 0.89 (12H, m) ppm. 2.2.3 (E)-6,6’-dibromoisoindigo thiophene (5) A solution of compound (4) (0.5 g, 0.75 mmol) and 2thiopheneboronic acid (0.048 g, 0.38 mmol) were dissolved in THF 20 ml. Catalyzed Pd2(PPh3)4 (0.021 g, 0.02 mmol). 2M Na2CO3 2.58 ml as base were added into solution. The reaction was refluxed for 24 h under N2 atmosphere. The solution was cooled and poured into water. The organic layer was collected and dried over Na2SO4. The crude product was purified by chromatography with hexane and dichloromethane (2:1) as eluent to give red-deep solid as 51% yield. 1H NMR (500 mHz, CDCl3) δ = 9.13 (1H, d, J=5 Hz), 9.02 (1H, d J=10.0 Hz), 7.41 (1H, d, J=5.0 Hz), 7.36 (1H,d, J=5.0 Hz) 7.28 (1H, d, J=5.0), 7.16 (2H, d, J=0 Hz), 7.11 (2H, m), 6.95 (1H, s), 6.87 (1H, s), 3.63 (4H, m), 1.83 (2H, t), 1.13 (16H, m) and 0.92 (12H, m) ppm. 2.2.4 IDTA A solution of compound (5) (0.17 g, 2.64 mmol), anthracene-9,10-diboronic acid bis(pinacol) ester (0.05 g, 2.12 mmol) was dissolved in THF 20 ml. Cs2CO3 (0.39 g, 1.2 mmol) and Pd2(PPh3)4 (0.014 g, 0.012 mmol) were added. . The reaction was refluxed for 24 h under N2 atmosphere. The solution was cooled and poured into water. The organic layer was collected and dried over Na2SO4. The crude product was purified by column chromatography with hexane and dichloromethane (2:1) as eluent to give black solid as 36% yield. 1H NMR (500 mHz, CDCl3) δ = 9.17 (4H, d, J=10.0 Hz), 8.34 (2H, d, J=5.0 Hz), 7.82 (2H, d,J=5.0 Hz), 7.44 (3H, s), 7.38 (4H, d), 7.32 (3H, d, Pure and Applied Chemistry International Conference 2015 (PACCON2015) 268 J=5.0 Hz), 7.14 (4H, d, J=5.0 Hz), 7.01 (4H, s), 3.72 (8H, m), 1.90 (4H. s), 1.28 (32H, m) and 0.92 (24H, m) ppm13C NMR (500 mHz, CDCl3) δ = 168.6 (4xC=O), 145.7 and 144.1 (Cq), 137.8 (Cq), 134.1 (Cq), 132.0 (Cq), 130.2 (CH), 128.3 (CH), 127.2 (CH), 126.1 (CH), 124.2 (CH), 121.0 (Cq), 119.3 (CH), 44.1 (CH2), 37.7 (CH), 30.8 (CH2), 29.7 (CH2) and 28.88 (CH2), m/z MALDI-TOF 1310.67 119.4 (CH), 44.1 (CH2), 37.1 (CH), 31.9 (CH2), 29.7 (CH2), 23.8 (CH2), 14.1 (CH3), 11.1 (CH3), m/z MALDI-TOF 1268.60 2.2.2.6 IDTF In 50 mL two-necked flask, (5) (0.12g, 0.2 mmol) and 9,9-dihexylfluorene-2,7-diboronic acid (0.05 g, 0.09 mmol), Pd(PPh3)4 (0.01 g, 0.009 mmol) as catalyst Cs2CO3 (0.29 g, 0.9 mmol) as base were dissolved in THF 20 ml. The mixture was refluxed for 24 h under nitrogen atmosphere. The reaction mixture was cooled and added 20 ml of water. The reaction was extracted with CH2Cl2 and washed with water, dried over anhydrous Na2SO4. Crude product was purified by column chromatography with hexane and dichloromethane (2:1) as eluent to give black solid as 54% yield. 1H NMR (500 mHz, CDCl3) δ = 9.24 (4H, d, J=10.0 Hz), 7.51 (4H, s), 7.45 (4H, d, J=5.0 Hz), 7.41 (4H, d), 7.22 (4H, s), 7.09 (4H, s), 1.98 (4H, s), 1.49 (40H, m) and 1.04 (30H, m) ppm.13C NMR (500 mHz, CDCl3) δ =168.6 (4xC=O), 145.7 (Cq), 144.1 (Cq), 137.8 (Cq), 132.0 (Cq), 130.2 (Cq), 128.2 (CH), 126.1 (CH), 124.2 (CH), 121.0 (Cq), 44.1 (CH2), 37.8 (CH), 30.8 (CH2), 29.7 (CH2), 28.8 (CH2), 24.2 (CH2), 23.0 (CH2), 14.1 (CH3), 10.8 (CH3), m/z MALDI-TOF 1466.86 2.2.5 IDTB A mixture of compound (5) (0.14 g, 0.22 mmol) and 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester) (0.04 g, 0.1 mmol) were dissolved in 20 mL of THF. Pd(PPh3)4 (0.012 g, 0.01 mmol) and Cs2CO3 (0.33 g, 1.0 mmol) were added into reaction and refluxed for 24 h under N2 atmosphere. The solution was cooled and poured into water. The solution was extracted with CH2Cl2. The combined organic layers were washed with water, dried over anhydrous Na2SO4. Crude product was purified using column chromatography with hexane and dichloromethane (2:1) as eluent gave black solid as 32% yield. 1H NMR (500 mHz, CDCl3) δ = 9.25 (4H, d, J=10.0 Hz), 7.51 (4H, d, J=5.0 Hz), 7.45 (4H, d, J=5.0 Hz), 7.39 (4H, d, J=10.0 Hz), 7.22 (4H, t), 7.09 (4H, s), 3.82 (8H, m), 1.98 (4H, s), 1.50 (32H, m) and 0.99 (24H, tt) ppm. 13 C NMR (500 mHz, CDCl3) δ = 168.6 (C=O), 145.7 (Cq), 144.1 (Cq), 137.7 (Cq), 132.0 (Cq), 130.2 (CH), 128.3 (Cq), 126.1 (CH), 124.2 (CH), 123.9 (CH), Br Br Br NH O O CH3COOH (2) O K2CO3 DMF O 96 % O N Br O conc. HCl NH + (1) H N Br N H (3) Br O N 84% S O O N O O Ar 51% O B O S N N 2M Na2CO3 Pd(PPh3)4 THF B(OH)2 Ar N S O B Br (4) S N O O Cs2CO3, Pd(PPh3)4 THF O N Br (5) Ar = N S N IDTA IDTB IDTF 36% 32% 54% Figure 1. Synthesis pathway of IDTA, IDTB and IDTF 3. Resultsand Discussion 3.1 Synthesis and Characterization The synthesis of three organic photovoltaic materials is showed in Figure 1. The (E)-6,6'-dibromoisoindigo (3) was synthesized by the presence of acid-catalyst aldol condensation of 6-bromooxindole and 6- bromoisatin in acetic acid[1]. The crude product was purified by column chromatography to give brown solid. Then alkylation reaction of 6,6’dibromoisoindigo using 1-bromo-2-ethylhexane in DMF give isoindigo derivatives (4)[1]. 1H NMR showed H of alkyl group near nitrogen atom at chemical shift 3.62 ppm (4H) and 1.82-0.89 ppm. Isoindigo derivatives were attached by Suzuki cross Pure and Applied Chemistry International Conference 2015 (PACCON2015) 269 in Figure 3 showed 3o amine stretching vibration was observed in the region 3500 cm-1which was the characteristic of lactam. C=O stretching vibration is improved at 1500 cm-1. IDTA IDTB IDTF 80 70 60 Transmittance coupling reaction of (4) and 2-thiopheneboronic acid in the presence of Pd(PPh3)4 as catalyst in THF to give (5). The target molecules were obtained by Suzuki cross coupling reaction of (5) and aryl diboronic ester in THF the presence of Pd(PPh3)4 as catalyst and Cs2CO3 as base. The 1H NMR spectra of IDTA, IDTB and IDTF showed in Figure 2. 1H-NMR spectrum of IDTA showed doublet signal at chemical shift 9.17 ppm (2H) assigning of H atom which carbonyl group. 8.35-1.01 ppm showed the aromatic proton. Multiplet peak at 3.72 ppm (8H) showed protons of alkyl. 1H NMR spectrum of IDTB showed doublet at 9.25 ppm (2H) which indicated the signal aromatic protons of Hbond and carbonyl group. Chemical shift 7.09-7.52 ppm showed aromatic protons. Chemical shift 3.82 ppm (8H) protons of alkyl groups near nitrogen atom.1H NMR spectrum of IDTF showed doublet signal at chemical shift 9.24 ppm (2H) proton assigning as aromatic which is bonding with carbonyl groups. Chemical shift 7.09-7.51 ppm was aromatic proton. Chemical shift 3.81 ppm (8H) showed multiplet which protons of alkyl near nitrogen atom and chemical shift 1-1.98 ppm as proton of alkyl chains. 50 40 30 20 C=O 10 o 0 4000 3 N-C 3500 3000 2500 2000 1500 1000 500 wavenumber (cm-1) Figure 3. IR spectra of IDTA, IDTB and IDTF 3.2 Optical properties The UV-Vis absorption spectra of organic photovoltaic materials in dilute CH2Cl2 solution are shown in Figure 4. The absorption spectra of all organic photovoltaic materials showed relative large molar extinction coefficient in visible region (300-600 nm). IDTF showed highest molar absorptivity relative to IDTB and IDTA. The synthesized molecules exhibited very low photoluminescence indicating these molecules can be used as donor materials in OPVs. 80000 IDTA IDTF IDTB 70000 (a) molar absorptivity 60000 50000 40000 30000 20000 10000 0 300 400 500 600 700 800 wavelength (nm) (b) Figure 4. Absorption spectra of IDTA, IDTB and IDTF in CH2Cl2 solution. 4. Conclusions In summary, we have successfully synthesized three organic photovoltaic materials which contained isoindigo bearing various core moieties (anthracene, benzothiadiazole and fluorene) as electron donors. The organic photovoltaic materials showed visible region (300-600 nm) in UV-Vis absorption spectra. (c) Figure 2. H NMR spectra of (a) IDTA, (b) IDTB and (c) IDTF The 13C NMR of IDTA, IDTB and IDTF showed chemical shift at 168.66 ppm assigning of carbonyl groups. The FT-IR spectra of IDTA, IDTB and IDTF 1 Acknowledgements This work was financially supported by Suranaree University of Technology. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 270 References [1] Christos, P., Xaver, P. and Helv, B.,1998,Chim Acta., 71, 1079–1083. [2] Wang, D., Ying, W., Zhang, X., Hu, Y., Wu, W. and Hua J., 2015 Dyes Pigm., 112, 327-334. [3] Wang, G., Tan, H., Zhang, Y., Wu, Y., Hu, Z., Yu, G. and Pan, C., 2014, Synth. Met., 187, 17-23. [4] Zhao, N., Qiu, L., Wang, X., An, Z. and Wan, X., 2014, Tetrehedron Lett., 1040-1044. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 271 SYNTHESIS TOWARDS N-HETEROAROMATIC THENA DERIVATIVE: INVESTIGATION ON [4+2]-CYCLOADDITION OF HIGHLY REACTIVE PYRIDYNE Chalupat Jindakun, Thanawon Chaisantikulwat, Jakapun Soponpong, Kulvadee Dolsophon, Tienthong Thongpanchang* Department of Chemistry, Faculty of Science, Mahidol University, Bangkok 10400, Thailand *E-mail: tienthong.tho@mahidol.ac.th Tel. +66 2201 5130, Fax. +66 2201 5139 Abstract: Analogous to 1,2,3,4-tetrahydro-1,4-epoxy naphthalene-1-carboxylic acid (THENA), a novel chiral derivatizing agent (N-THENA 4) bearing a pyridine moiety as an anisotropic group was first designed and synthesized with the main objective to study the anisotropic effect of heteroaromatic moiety in the absolute configuration analysis. The key synthetic step is the formation of bicyclic compound 3, which was directly synthesized from [4+2]-cycloaddition reaction of a highly reactive 2,3-pyridyne intermediate 1 and an electron deficient methyl furan-2-carboxylate 2. A variety of conditions to generate 2,3-pyridyne intermediate from 3(trimethylsilyl)pyridin-2-yl triflate were investigated and the bicyclic compound 3 was prepared in 5% yield under the optimal condition. O O CO2 Me + 1 2 O [4+2] cycloaddition N At present, there are a number of common CDAs generally used to determine the configuration of chiral compounds such as -methoxy--trifluoromethyl-phenylacetic acid (MTPA) or acid chloride derivative (MTPACl), both of which are usually called Mosher’s reagents[2a‒b], -methoxy--phenylacetic acid (MPA)[2b‒c] and -(9-anthryl)--methoxyacetic acid (9-AMA)[3] (Figure 2). Their conformations are presumably controlled by the stereoelectronic effect of elecron-withdrawing groups (trifluoromethyl or methoxy groups) and steric effect of the aryl ring, which orient high electronegative group syn-periplanar to the carbonyl group[4]. steps CO 2Me 3 N CO 2H N N-THENA 4 1. Introduction Determining the absolute configuration of chiral compounds is essential for natural products and asymmetric syntheses. There are several instrumental methods to define the absolute configuration such as chiroptical methods[1a‒b], X-ray crystallography [1c‒d] or chiral HPLC[1e‒g]. NMR spectroscopy is reportedly a powerful technique due to its convenience and no reference standard requirement[1h‒i]. To determine the configuration via the NMR technique, chiral compounds with undefined stereochemistry are derivatized with a pair of enantiomers of a chiral derivatizing agent (CDA) to generate two diastereomeric forms. As a result of the anisotropic effect of the aromatic moiety of CDA, 1H-NMR spectra of the diastereomers display the distinct signals of which the differences (L1 and L2) could indicate the absolute configuration of chiral compounds (Figure 1). Figure 2. Common chiral derivatizing agents Recently, our group has synthesized a novel CDA, tetrahydro-1,4-epoxynaphthalene-1-carboxylic acid (THENA), for defining the chirality of secondary alcohols (Figure 3)[5]. The conformation of THENA was locked with covalent bonds in a bicyclic form, resulting in only the deshielding effect to play a vital role in differentiating spectroscopic signals. Although most CDAs depend on the anisotropic effect of the aromatic hydrocarbon, the effect of heteroaromatic moiety has never been investigated. Based on the rigid structure of THENA, the new CDA bearing pyridine ring (N-THENA 4) is designed and synthesized to study the effect of pyridine moiety in configuration analysis (Figure 3). O CO 2H THENA O CO2 H N N-THENA 4 Figure 3. New chiral derivatizing agent bearing N-heteroaromatic moiety (N-THENA 4). Figure 1. Concept of chiral derivatizing agent (CDA) for determining the absolute configuration by NMR spectroscopy. Relating to the synthesis of THENA, the retrosynthetic analysis of N-THENA 4 was shown in Scheme 1. The desired target molecule 4 can be Pure and Applied Chemistry International Conference 2015 (PACCON2015) 272 achieved by hydrolysis and hydrogenation of the Diels-Alder adduct 3, which could be obtained from [4+2]-cycloaddition of 2,3-pyridyne 1 and methyl furan-2-carboxylate 2. Apparently, the Diels-Alder reaction of 2,3-pyridyne 1 with furan carboxylate 2 is of great challenge due to the electron deficient nature of the diene. Diels-Alder O O hydrolysis CO2H N N-THENA (4) and hydrogenation O CO2Me N 3 CO2Me + N 1 2 Scheme 1. The retrosynthetic analysis of N-THENA 4 Reportedly, the pyridyne intermediate could be generated from two types of precursors[6‒7] as depicted in Scheme 2. A sequential halogen-metal -elimination of 3-bromo-2exchange and chloropyridine 6 with strong base (Method A) yielded the 2,3-pyridyne intermediate 1[6a]. An alternative milder condition was fluoride-induced desilylationelimintation of 3-(trimethylsilyl)pyridin-2-yl triflate 7 with anhydrous CsF (Method B) which also generated the desired 2,3-pyridyne 1 [7]. Scheme 2. Two reported methods for 2,3-pyridyne intermediate formation. 2. Materials and Methods 2.1 General Experiment Merck silica gel 60H was used for column chromatography. 1H-NMR spectra were recorded on Bruker Ascend 400 MHz spectrometer in deuterated solvents. All reactions were conducted in oven-dried glasswares, unless otherwise instructed in the procedure. All solvents were distilled before use. In case of tetrahydrofuran (THF), it was freshly distilled over sodium/benzophenone under nitrogen atmosphere. 2.2 Synthetic Method 2.2.1 Synthesis of methyl 5,8-dihydro-5,8-epoxy quinoline-8-carboxylate 3 Method A: The method was modified from that reported in ref.[6a]. A 1.6 N solution of n-butyllithium in hexane (0.71 mL, 1.14 mmol) was added dropwise to a solution of 3-bromo-2-chloropyridine 5 (200 mg, 1.04 mmol) in THF (3 mL) at ‒78oC over 15 min under nitrogen atmosphere and left stirring for 30 min. Then methyl furan-2-carboxylate 2 (0.9 mL, 8.41 mmol) was slowly dropped to the reaction mixture at ‒78oC over 5-10 min, and the reaction mixture was allowed to stir at room temperature for 1 h. After that, the reaction was quenched with water and extracted with dichloromethane (3x20 mL). All organic layers were combined, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography with 1:6:13 MeOH:EtOAc:hexane as an eluent to afford 54 mg (25 %) of (2-chloropyridin-3-yl)(furan-2-yl)methanone 8 as a brown solid and 53 mg (16%) of (2-chloropyridin3-yl)di(furan-2-yl)methanol 9 as a brown solid 1 H NMR of 8 (CDCl3) δ: 8.51 (dd, 3J = 4.8 Hz, 4J = 1.9 Hz, 1H), 7.79 (dd, 3J = 7.5 Hz, 4J = 1.9 Hz, 1H), 7.69 (d, 3J = 0.9 Hz, 1H), 7.36 (dd, 3J = 4.8 Hz, 7.52 Hz, 1H), 7.12 (d, 3J = 3.6 Hz, 1H), 6.59 (dd, 3J = 1.6 Hz, 3.6 Hz, 1H). 13C NMR (CDCl3) δ: 181.1, 152.5, 151.9, 149.3, 148.8, 138.8, 134.6, 122.8, 122.3, 113.6. 1 H NMR of 9 (CDCl3) δ: 8.39 (dd, 3J = 4.5 Hz, 4J = 1.5 Hz, 2H), 7.73 (dd, 3J = 7.9 Hz, 4J = 1.7 Hz, 2H), 7.51 (d, 3J = 1.1 Hz, 1H), 7.28 (dd, 3J = 4.7 Hz, 7.9 Hz, 2H), 6.42 (dd, 3J = 3.3 Hz, 4J = 1.4 Hz, 1H), 6.20 (d 3J = 3.3 Hz, 1H). 13C NMR (CDCl3) δ: 152.8, 149.2, 148.8, 143.2, 139.6, 136.9, 122.3, 111.0, 110.9, 76.6. Method B: A mixture of methyl furan-2carboxylate 2 (2 mL, 18.7 mmol) and 3(trimethylsilyl) pyridin-2-yl triflate 7 (500 mg, 1.67 mmol)[7] was slowly transferred to flame-dried cesium fluoride at room temperature under nitrogen atmosphere. The reaction was allowed to stir vigorously at ambient temperature for 3 h. Then the reaction mixture was diluted with EtOAc (20 ml) and washed with water twice. The aqueous layer was extracted back with EtOAc twice and the organic layers were combined, dried with Na2SO4 and concentrated in vacuo. The excess furan ester 2 was removed by Kugelrohr distillation under reduced pressure and the residue was purified by flash column chromatography using 1:6:13 MeOH:EtOAc:hexane as an eluent to afford methyl 5,8-dihydro-5,8-epoxyquinoline-8-carboxylate 3 as a yellow solid in 8.4 mg (5.1 %yield) 1 H NMR of 3 (MeOD-d4) δ: 7.97 (d, 3J = 5.3 Hz, 1H), 7.65 (d, 3J = 7.2 Hz, 1H), 7.23‒7.19 (m, 2H), 7.03 (dd, 3J = 5.4 Hz, 7.2 Hz, 1H) 5.94 (d, 3J = 1.48 Hz, 1H), 3.97 (s, 3H). 13C NMR (MeOD-d4) δ: 172.1, 168.1, 146.3, 144.1, 143.4, 141.8, 135.8, 121.6, 91.8, 83.1, 53.4. 2.2.2 Synthesis of methyl 5,6,7,8-tetrahydro-5,8-epoxy quinoline-8-carboxylate 12 A mixture of compound 12 (69 mg, 0.34 mmol) and 10% Pd/C (7 mg) in methanol (5 mL) was allowed to stir at room temperature for 16 h under hydrogen atmosphere. The reaction mixture was filtered through a pack of Celite pad and washed with MeOH. The filtrate was then concentrated in vacuo to give 70 mg (quantitative) of the desired product as yellow oil. The crude product was used in the next step without further purification. 1 H NMR of 12 (MeOD-d4) δ: 8.25 (dd, 3J = 5.2 Hz, 4 J = 1.3 Hz, 1H), 7.76 (dd, 3J = 7.4 Hz,4J = 1.3 Hz, 1H), 7.27 (dd, 3J = 5.3 Hz, 7.5 Hz 1H), 5.59 (d, 3J = 4.4 Hz, 1H), 3.94 (s, 1H), 2.31‒2.21 (m, 2H), 1.78‒1.73 (m, 1H), 1.55‒1.50 (m, 1H). 13C NMR (MeOD-d4) δ: 169.9, 165.2, 147.5, 140.2, 129.2, 123.9, 88.0, 79.5, 53.2, 29.3, 28.7. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 273 2.2.3 Synthesis of 5,6,7,8-tetrahydro-5,8-epoxy quinoline-8-carboxylic acid (N-THENA) 4 Pulverized potassium hydroxide (95 mg, 1.7 mmol) was added to a solution of 12 (70 mg, 0.34 mmol) in 1,4-dioxane and MeOH (1:1, 0.6 mL). Then the reaction was allowed to stir at room temperature for 3 h. The reaction mixture was diluted with MeOH and acidified with 1 N HCl to pH 1-2 at room temperature and evaporated to dryness. The crude product was desalinated by adding a mixture of EtOAc/MeOH (1:1) and filtration. The mother liquor was evaporated and the obtained solid was recrystallized in hot acetone to give 71 mg (75% yield) of N-THENA as a white crystal. 1 H NMR of 4 (MeOD-d4) δ: 8.23 (d, 3J = 4.8 Hz, 1H), 7.66 (d, 3J = 7.3 Hz, 1H), 7.16 (dd, 3J = 5.5 Hz, 7.1 Hz 1H), 5.46 (d, 3J = 4.9 Hz, 1H), 2.25‒2.20 (m, 1H), 2.16‒2.12 (m, 1H), 1.77‒1.71 (m, 1H), 1.48‒1.44 (m, 1H). 13C NMR (MeOD-d4) δ: 170.6, 163.3, 145.2, 142.0, 131.8, 124.8, 87.6, 79.5, 29.8, 28.5. From Table 1, treatment of acetonitrile solution of pyridyne precursor 6 with CsF in the presence of furan ester 2 gave only a trace of Diels-Alder adduct 3 (entry 1). An increased amount of 2 under neat condition gave a better yield of the desired product 3 as expected (entries 2 and 3). In addition, flame-dried CsF under vaccum gave a better yield than normal dried CsF under vaccum (entries 5 and 6). This may be due to the presence of water in the normal dried CsF. Table 1: Diels-Alder reaction condition of 6 with 2a Fsource furan 2 (equiv) 1 CsF 4.0 2 CsF 3. Results and Discussion 3 A halogen-metal exchange-elimination sequence of dihalopyridine 5 with strong base (Method A) was first carried out. In this study, no Diels-Alder adduct was observed. Instead, our result indicated that the lithiated pyridine 7 underwent 1,2-addition with furan carboxylate 2 to yield ketone 8 and alcohol 9 (Scheme 3). This observation suggested that, under our reaction condition, the 1,2-addition (pathway B) of pyridyl lithium 8 to carbonyl group is preferable than the 1,2elimination (pathway A) to form pyridyne intermediate[6]. 4 pathway A N 1 Br Li n-BuLi N 5 Cl THF, 78 oC 30 min N Cl 7 N O O pathway B MeO 2C O N + Cl OH Cl N 2 78oC to rt 8 (25%) Cl O 9 (16%) Scheme 3. Two possible competitive reactions of (2bromopyridin-3-yl)lithium 7, trapping with furan 2. The mild and effective method for in situ pyridyne formation was achieved from treatment of 3(trimethylsilyl)pyridin-2-yl triflate 6 with anhydrous CsF[7]. Triflate 6 was synthesized from 2hydroxypyridine 11 in high yield over 2 steps (Scheme 4)[7b]. Scheme 4. Synthetic route of a pyridyne precursor 6. Entry Time (h) Yieldb (%) 18 0.7 4.0 Solvent (mL) MeCN (1.7 mL) neat 18 2.4 CsF 11.1 neat 18 4.1 CsF 11.1 neat 1 1.7 5 c CsF 11.1 neat 1 4.1 6c CsF 11.1 neat 3 5.3 7 TBAF 1M TBAF 11.1 neat 3 2.3 11.1 THFd 18 0 8 a All reaction using 1.67 mmol of 6 reacting with 4 equiv of Fsource at ambient temperature under neat condition. b isolated yield of 3. c Anhydrous CsF was dried with Bunsen-flame under vacuum prior to use. d solvent was from 1M TBAF in THF. Tetrabutylammonium fluoride (TBAF), consisting of more hydrophobic moiety, was used to allow the reaction to perform as a homogeneous solution (entry 7). The desired product, however, was produced in lower yield. Additionally, when commercially available 1M solution of TBAF in THF was used, the desired product cannot be detected (entry 8). It is possible that moisture in the commercial reagent could potentially impede the reaction[7a]. The position of nitrogen atom in 3 was confirmed by 2D NOESY experiment. The result showed a crossrelaxation peak between H-4 ( = 7.65 ppm) and the bridgehead proton (H-b, = 5.95 ppm) as shown in Figure 4. Another adduct with different regiochemistry could also be formed. However, the pure compound could not be isolated from other uncharacterized byproducts. Figure 4. Key NOESY cross relaxation peak of 3. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 274 The unsaturated bicyclic 3 was then reduced by catalytic hydrogenation to provide the saturated bicyclic ester 5 in quantitative yield. In the final step, hydrolysis of ester 5 was conducted by potassium hydroxide in a mixture of dioxane and methanol to obtain the target product in 75% yield (Scheme 5). [5] [6] [7] Scheme 5. Reactions of 3 toward N-THENA 4 by catalytic hydrogenation and subsequent hydrolysis. Sungsuwan, S., Ruangsupapichart, N., Prabpai, S., Kongsaeree, P. and Thongpanchang, T., 2010, Tetrahedron Lett., 51, 4965‒4967. (a) Cook, J. D. and Wakefield, B. J., 1969, J. Chem. Soc. C, 1973‒1978. (b) Walter, M. A. and Shay, J. J., 1995, Tetrahedron Lett., 36, 7575‒7578. (c) Connon, S. J. and Hegarty A. F., 2001, Tetrahedron, 42, 735‒737. (d) Connon, S. J. and Hegarty, A. F., 2004, Eur. J. Org. Chem., 3477‒3483 (a) Walters, M. A. and Shay, J., 1997, Syn. Comm., 27, 3573‒3579. (b) Carroll, F. I, Robinson, T. P., Brieaddy, L. E., Atkinson, R. N., Mascarella, S. W., Damaj, M. I., Martin, B. R. and Navarro, H. A., 2007, J. Med. Chem., 50, 6383‒6391. (c) Effenberger, F. and Daub, W., 1991, Chem. Ber., 124, 2119‒2125. 4. Conclusions A reaction of pyridyne and electron-deficient diene was first reported. It was found that 6 was the only pyridyne precursor able to provide the desired product 3 in 5% yield while 5 could not generate the 2,3pyridyne intermediate. The key compound 3 was subsequently reduced and hydrolyzed to afford NTHENA 4 in 75% yield over 2 steps. The structure of product was assured by NMR experiments. Acknowledgements Financial and facility support from Faculty of Science, Mahidol University and The Center of Excellent for Innovation in Chemistry (PERCH-CIC) are gratefully acknowledged. References [1] [2] [3] [4] Instrumental techniques for the determination of the absolute configuration: Chiral optical methods (a) Polavarapu, P. L., 2002, Chirality, 14, 768‒781 (b) Sundararaman, P., Barth, G. and Djerassi, C., 1981, J. Am. Chem. Soc., 103, 5004‒5007.; X-ray crystallography (c) Harada, N., 2008, Chirality, 20, 691‒723. (d) Bijuvoet, J. M., Peerdeman, A. F. and Bommel, A. J. 1951, Nature, 168, 271‒272.; Chiral HPLC (e) Roussel, C., del Rio, A., Pierrot-Sanders, J., Piras, P. and Vanthuyne, N., 2004, J. Chromatogr. A, 1037, 311‒328. (f) Kobayashi, J., Hosoyama, H., Katsui, T., Yoshida, N. and Shigemori, H., 1996, Tetrahedron, 52, 5391‒5396. (g) Luesch, H., Yoshida, W. Y., Moore, R. E. and Paul, V. J., 2000, J. Nat. Prod., 63, 1437‒1439.; and NMR (h) Seco, J. M., Quiñoá, E. and Riguera, R., 2004, Chem. Rev., 104, 17‒117. (i) Parker, D., 1991, Chem. Rev., 91, 1441‒1457. (a) Dale, J. A., Dull, D. L. and Mosher, H. S., 1969, J. Org. Chem., 34, 2543‒2549. (b) Dale, J. A. and Mosher, H. S., 1973, J. Am. Chem. Soc., 95, 512‒519. (c) Dale, J. A. and Mosher, H. S., 1968, J. Am. Chem. Soc., 90, 3732‒3738. Seco, J. M., Latypov, Sh. K., Quiñoá, E. and Riguera, R., 1994, Tetrahedron Lett., 35, 2921‒2924. (a) Latypov Sh. K., Seco, J. M., Quiñoá, E. and Riguera, R., 1995, J. Org. Chem., 60, 504‒515. (b) Seco, J. M., Latypov, Sh. K., Quiñoá, E. and Riguera, R., 1997, Tetrahedron, 53, 8541‒8564. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 275 CHEMOSELECTIVE REDUCTIONS OF NITROBENZENE USING CHITOSAN-COATED METAL AS A CATALYST Thamonwan Angkuratipakorn, Pawinna Chanthawong, Jirada Singkhonrat* Department of Chemistry, Faculty of Science and Technology, Thammasat University, KlongLaung, Pathumthani, 12120, Thailand *E-mail: jirada@tu.ac.th, Tel. +662 564440 ext 2417, Fax. +662 564 4483 Abstract: A micellar technology for chemoselective reductions has been developed using chitosan-coated metal as a catalyst. Our works focus, in part, on the design of nonionic surfactants that enable transitionmetal-catalyzed reactions to be performed in water at room temperature (rt), rather than in traditional organic solvents. Several applications of micellar technology to an array of valued organic transformations have already been developed. To further expand the scope of these micellar surfactant conditions, zinc mediated reductions of nitrobenzene offer 100% conversion within 2 hours at rt. functionalization. Herein, we attempt to develop a transition-metal-free approach for reduction of nitroarenes in H2O employing a modified chitosan as a hydrogen source (Scheme 1). The use of modified chitosan as reducing agent for transition metal-free reduction is inspiring. Therefore, carrying out the reaction in water without employing any metal or with low loading of metal could make the present method highly attractive. 1. Introduction 2.1 Chemicals All commercially available compounds were used as received. A representative hydrophobic nitrobenzene was examined in a 2% w/v of aqueous solution of the designed surfactant PGS oligomer, which is a bio-surfactant developed in our research group (Scheme 2). Addition of zinc dust and ammonium chloride to a solution of nitrobenzene 1 resulted in a clean conversion to aminobenzene 2 upon stirring for 2 h at rt (Scheme 1). Aromatic and heteroaromatic primary amines are important as building blocks for the synthesis of a wide range of pharmaceuticals, polymers, special and fine chemicals. While a wide range of protecting groups are available to bound amine reactivity, one popular approach involves their masking as nitroarenes. This approach offers multiple benefits, including facile nitrogen introduction via early stage nitration, tremendousatom economy, good stability of the nitro group under numerous reaction conditions, and directed arene functionalization resulting from the associated electronics of the nitromoiety[1]. Catalytic hydrogenation using transition metal catalysis has received high interest, but this approach exhibits limited selectivity in the presence of other reducible functional groups, similar to transfer hydrogenation processes which often show a decrease in yields in the presence of other reducible groups [2]. In addition, the major drawbacks linked to some of those conditions are the difficulty to handle reactants and the utilization of toxic or expensive reagents, for example, though the high toxicity, potential flammability, and instability of hydrazine. In the search of new methodologies to reduce organic molecules, the utility of micellar technology was studied. The micellar surfactant conditions have been already identified as efficient reactants to promote reductions in the presence of transition metals [3]. One of the major challenges in academia and industry is the development of environmentally friendly synthetic processes. This communication aims at developing an efficient method to reduce nitrobenzene selectively into aminobenzene employing micellar technology catalyzed by chitosan-coated Zn bead nanoparticles. Thus, selecting one method amongst the two specific conditions should provide a powerful and helpful toolbox for selective 2. Materials and Methods NO2 2 equiv. NH4Cl H2O NH2 4%wt PGS oligomers chitosan - Zn dust RT , 2h Nitrobenzene 1 100% conversion Aminobenzene 2 Scheme 1: Reduction of nitrobenzene in green solvents (water) at rt Monitoring the reaction products by GCMS (GCMS-QP2010 Ultra SHIMADZU using Column HP-5MS (30 m x 0.25 mm x 0.25 µm) with temperature limits from 60 to 325oC) revealed that no residual nitroso 3a-3c or hydroxylamine intermediates was found at full conversion (Table 1, entry 6). The amino product 2 in more than 95% yield was obtained by using simple workup including filtration through a silica gel plug followed by rinsing with a minimal volume of ethyl acetate and aqueous ammonium hydroxide. The organic layer was recovered by simple decantation, dried over MgSO4, filtered, and evaporated under reduced pressure. Alternatively, extraction with a minimal volume of organic solvent led to the isolated product with similar purity and yield. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 276 2.2 Modification of chitosan (CS) Modified chitosan was prepared as follows: Chitosan (CS, 0.4 g) was dissolved in distilled water (50 mL) 2% acetic acid with constant stirring for approximately 24 h at rt. The homogeneous solutions were poured and precipitated with bentonite solution to obtain CS-immobilized on bentonite (CIB). The bead polymers were allowed to dry at 60°C for 24 h. The dried chitosan matrix was heated at 100°C under moderate vacuum for 2 h. Modified chitosans were studied for their structural characteristics using X-ray Diffractometry (XRD) and Fourier Transform Infrared Spectroscopy (FTIR). Thermogravimetric analyses were performed with a Netzsch TG 209 C apparatus. Surface modification of Zn dust with chitosan (CS) Zn dust was used for coating procedure. 1% Bare Zn dust was dispersed in 0.5% and 1% (w/v) CS solution in 2% (v/v) acetic acid (1:1 and 1:2 of CS:Zn). The Zn dust was activated by 0.1M HCl. The mixture was ultrasonicated for 30 min. After ultrasonication, the CS-coated Zn dust (CS-Zn) was allowed to settle, and was washed with distilled water (3 times) in order to remove excess CS. These CS-Zn catalysts were then separated (wet CS coated-Zn) and dried at 50°C for 24 h (dried CS coated-Zn) used in the reaction as initial Zn weight for preparation above. The Catalysts are under investigation for their structural characteristics using X-ray Diffractometry (XRD), Fourier Transform Infrared Spectroscopy (FTIR), Thermo-Gravimetric Analysis (TGA), Scanning Electron Microscopy (SEM as showed in scheme 3), and Transmission Electron Microscopy (TEM). C14H29 O O O OH HO (b) Scheme 3. SEM micrographs of (a) Zn dust and (b) chitosan-coated Zn dust (dried CS coated-Zn) at 0.5 M substrate concentration. Low yield was observed in the reaction condition with unactivated Zn and with activated Zn under th e conventional workup (Table 1, entries 1-2 and entries 3-4, respectively). A suspension formed in the aqueous phase during the extraction with ethyl acetate was due to the inherent insolubility of the catalyst. OH O O (a) O 2 O O O O MW = 910 OH Scheme 2. Molecular structure of palmitoyl-(PGS)3glycerol (PG1.5SF0.32) [4] Procedure for Reduction of nitroarenes. To a solution of 4.87 mmol (0.5 M) of nitrobenzene in water (10 mL) were added 9.74 mmol (2 equiv) of NH4Cl, PG1.5SF0.32 4wt% and Zn dust 48.7 mmol (10 equiv). The mixture was stirred at room temperature for the desired time (Table 1). A simple workup entails filtration through a silica gel plug followed by rinsing with a minimal volume of ethyl acetate and ammonium hydroxide. The product was extracted with ethyl acetate. The ethyl acetate layer was evaporated at reduced pressure on a rotary evaporator. 3. Results and Discussion In the first set of the experiments, the reactivity of substrate 1 in aqueous micellar conditions was studied at rt in water. The nitrobenzene was a model substrate Scheme 4. Possible pathways for the reduction of nitrobenzene by Zn dust in PG1.5SF0.32/H2O and NH4Cl at rt Therefore, the reduced intermediates 3a-3c and reduced product 2 were trapped within micelle. The use of silica gel filtration was introduced for work-up step. For reduction in aqueous media, it was reasoned that micelles acted as carriers of the insoluble substrate from the solution to the catalyst surface and improved the diffusion to reactive centers. The reduced product 2 was then expelled from the micelles as its water Pure and Applied Chemistry International Conference 2015 (PACCON2015) 277 solubility was increased and extracted with ethyl acetate and ammonium hydroxide solution during work-up process to achieve higher yield (Table 1, entries 5-6). In the absence of added additives, the conversion of nitrobenzene 1 was quantitative when the reaction was carried out in water for 3 and 2 hours employing both 2%wt and 4%wt surfactants (PGS oligomer), respectively (Table 1, entries 5 and 6). Additionally, to investigate the reduction pathway, the reduction of nitrobenzene 1 was monitored by GCMS. Masses corresponding to the intermediates would be able to suggest transformation under which reduction conditions, i.e., direct or condensation pathway is carried out. When the reaction of nitrobenzene catalyzed by the Zn dust was performed successfully under the conditions described in Scheme 1, no traces of azoxybenzene 3a or diazene 3b were detected (GC-MS). When the reduction of nitrobenzene 1 was carried out under the optimum reaction conditions albeit with low Zn dust loading, no reduced product 2 was formed, but also provided reduced intermediate 3 (azoxybenzene 3a and diazene 3b) as the major products (Table 1, entries 7-8). Therefore, these results clearly suggested that the condensation route was predominant in the present reaction conditions (Scheme 4). Lowering the catalyst loading (5 equiv.) showed to be ineffective in terms of substrate conversion or yield of reduced product 2 (Table 1, entry 7) and revealed no effect or improvement from higher amount of the surfactant (Table 1, entry 8). Problems of azoxybenzene 3a accumulation are more frequently encountered for low catalyst loading due to the low reduction capacity. The reactivity of Zn was inhibited by the presence of hydrophobic surrounding protecting the intermediate 3a from the formation of intermediate 3b as observed for reduction in low catalyst loading (5 equiv.). The Zn dust cannot be reuse in this study as indicated by the low reduction power to achieve intermediates 3a-3b (Table 1, entry 9). The results using two different additives (NH4Cl and N2H4.H2O; Table 1, entry 5 and entry 10, respectively) were also compared in water micellar conditions at rt. Lower hydrogen transfer ability of hydrazine was found in this this study for the optimum condition with high composition of diazene 3b and low conversion of 19% to the reduced products 2. Table 1 : Reduction of nitrobenzenea (Optimization of zinc-mediated reductions) Condition Untreated Zn with Conventional Entry 1 2 Zn dust (equiv) 10 5 PG1.5SF0.32 (wt%) 2 2 Additives Time (h) 6 4,6 1 - 3a - %conversion* 3b - 2 - 10 2 6 0.2% 0.3% 99.5% 10 2 2 10 2 3 100% NH Cl 4 10 4 2 100% 5 2 3 73% 27% 5 4 3 Activated 10b 4 2 8% 89% 3% Zn with (Reused) silica plug 10 c 10 2 N2H4.H2O 3 1% 72% 8% 19% work-up 11d 10 2 CIB/water 6 86% 7% 7% 12e 10 CIB/methanol 3 13f 10 CIAB/methanol 3 96% 4% 14g 5 4 CIB/water 2 91.5% 8.5% 15h 10 4 NH4Cl 2 32% 24% 42% 2% Wet CS-Zn (1:1) 16i 10 4 NH4Cl 2 0.4% 3% 96.6% Dried CS-Zn (1:2) Conditions at room temperature with *GC Analysis: a0.5M nitrobenzene, PG1.5SF0.32/H2O, NH4Cl 2 equiv .bReused Zn from entry 8 c Replaced NH4Cl with N2H4.H2O 2 equiv.dUse of chitosan immobilized on bentonite (CIB) as a hydrogen source and a reducing agent in water e similar to entry 11 in methanol fUse of chitosan immobilized on activated bentonite (CIAB) in methanol gPG1.5SF0.32/H2O, NH4Cl 2 equiv and CIB 1 gram. hWet CS-coated Zn dust in PG1.5SF0.32/H2O, NH4Cl 2 equiv. iDried CS-coated Zn dust in PG1.5SF0.32/H2O, NH4Cl 2 equiv. work-up 3 4 5 6 7 8 9 3c - Pure and Applied Chemistry International Conference 2015 (PACCON2015) 278 Compared to its unstable anhydrous form, hydrazine hydrate is relatively safe and easy to handle and was applied to this study. In the past few years, several protocols for the reduction of nitroarenes employing a combination of hydrazine and iron catalysts have been reported at higher temperature. Herein, hydrazine-Zn mediated protocols showed low conversion leading to mainly diazene 3b, and revealed hydrazine 3c (Table 1, entry 10). Other additives such as chitosan were applied (Table 1, entries 11-15). The use of modified chitosan as a hydrogen source and reducing agent for transition metal-free reduction is introduced. Beads of modified chitosans were immobilized on bentonite directly and performed in the reaction (Table 1, entries 11-13). The thermal stability of native chitosan (CS sample), chitosan bead (CSbead) and chitosan immobilized on bentonite (CIB sample) was studied by TGA. Two major weight-loss stages at approximately 40–250°C and 250–670°C were observed in the tested temperature range. The first weight loss at approximately 40–250°C is due to the volatility of small molecules, such as physically absorbed water and acetic acid. The second weight loss is due to the degradation of CS, CS-bead and CIB catalyst, which can be assigned to the structural collapse of the chitosan and the thermal decomposition of the polymeric network. The chitosan immobilized on bentonite (CIB sample) showed significant improvement in the thermal stability. The CIB catalyst is therefore stable and selected at the desired operating condition. It exhibited to lower reducing power of Zn dust and gave intermediate 3b with insufficient hydrogen source to reduced product 2 (Table 1, entry 11). To observe the reaction conditions of CIB catalyst, the reduction of nitrobenzene 1 without micellar surfactant was carried out in the presence of methanol. No reaction was occurred (Table 1, entry 12). The procedure for activated bentonite was then carried out to prepare the CIB and was used in the reaction in the absence of micellar surfactant (Table 1, entry 13) obtaining similar result as entry 11 with azoxybenzene 3a accumulation. Poor results were observed with the attempt to improve reducing power by using Zn dust based on micellar technology (Table 1, entry 14). Finally, in order to increase the reducing power and offer higher ability to transfer hydrogen, CS-coated Zn dust was employed as initially prepared in Zn equiv. under the optimized conditions (Table 1, entry 6). The intermediates 3a-3c were observed i9n higher hydrogen transfer ability for wet CS coated-Zn (Table 1, entry 15). The performance of dried CS-coated Zn dust as potential reducing agent with good hydrogen source were achieved (Table 1, entry 16).The ability of the CS coated-Zn as reusable materials based on micellar technology is in progress. stirring for 3 hours. The break-through reported in this paper aims at developing a commutative strategy based on micellar technology using PGS oligomer as nonionic surfactants and CS coated Zn as catalyst for reducing agent. Further studies on the modified chitosan in catalytic application are being carried out in our laboratory. Acknowledgements The authors are thankful to Department of Chemistry, Faculty of Science and Technology, Thammasat University.The authors also gratefully acknowledge the partial support provided by Central Scientific Instrument center (CSIC) Faculty of Science and Technology Thammasat University. Contract No 001/2556. References 1. Lipshutz, B. H., Ghorai, S., Abela, A. R., Moser, R., Nishikata,T., Duplais, C., Krasovskiv, A., Gaston, R. D. and Gadwood, R. C., 2011, J. Org. Chem., 76, 43794391. 2. (a) Sheldon, R. A., 2007, Green Chem., 9, 1273. (b) Lipshutz, B. H., Isley, N. A., Fennewald, J. C. and Slack, E. D., 2013, Angew. Chem., Int. Ed., 52, 10952-10958. 3. Kelly, S.M. and Lipshutz, B. H., 2014, Org. Lett., 16, 98101. 4. Khongphow C., Theerakul, J., Puttamat, S. and Singkhonrat, J., 2015, Characterization of poly(glycerolsuccinate) oligomers as bio-based non-ionic surfactants by nuclear magnetic resonance and mass spectrometry, Colloids Surf. A: Physicochem. Eng Asp., 468, 301-308. 4. Conclusions In summary, the optimized conditions were set at 0.5 M nitrobenzene, 10 equiv. of fresh zinc dust, 2%wt PGS oligomer and 2 equiv. of ammonium chloride at rt Pure and Applied Chemistry International Conference 2015 (PACCON2015) 279 SYNTHESIS AND CHARACTERIZATION OF CYANURIC ACID SUBSTITUTED 1,8-NAPHTHALIMIDES Chittranuch Pengsawad1, Mongkol Sukwattanasinitt2, Paitoon Rashatasakhon2* 1 Program in Petrochemistry and Polymer Science, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand 2 Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand *E-mail: paitoon.r@chula.ac.th Tel. +66 2218 7596, Fax +66 2218 7598 Abstract: Two novel fluorescent sensors (1 and 2) are designed as potential sensors for melamine. They are successfully synthesized from 1,8-naphthalimide as fluorophore and cyanuric acid as receptor for melamine. The synthesis involves an imidation of 1,8-naphthalic anhydride with ethanolamine, transformation of the hydroxyl group to a bromo group, and N-alkylation of isocyanuric acid by the bromo group. The target compounds are characterized by 1H-NMR, 13C-NMR, mass spectroscopy, and elemental analysis. The photophysical properties are investigated by UV-Vis and fluorescence spectroscopy. The difference in substitution at the 4-position of the naphthalimide moiety plays an important effect on the emission wavelengths and fluorescence efficiencies. Progress on the optimization of a sensing system for melamine will be presented. 1. Introduction Melamine (C3H6N6) is a high nitrogen-containing compound (66% by mass) which is essential for the production of melamine-formaldehyde resins used in plastic, paints, adhesives, and fire retardant. Recently, melamine was contaminated in milk product and infant formula for the false increase of protein levels as determined by the nitrogen contents. Although melamine has low toxicity, it can be associated with cyanuric acid to from high molecular weight network complexes. The complex has poor aqueous solubility and precipitates in renal tubes, causing damage to the urinary system developing kidney stones, and ultimate death [1]. Therefore, The U.S. Food and Drug Administration (FDA) set a safe contamination level of melamine at 1 mg kg-1 for powdered infant milk formula and at 2.5 mg kg-1 for other foods [2]. Nowadays, several methods for melamine detection [3] have been reported such as gas chromatography/mass spectrometry (GC-MS) [4], high-performance liquid chromatography (HPLC) [5], liquid chromatography (LC) [6], and enzyme-linked immunosorbent assay (ELISA) [7]. Most of these methods are highly sensitive, but they require highcost instruments, complicated sample preparation, and well-trained technicians or instrument users. Therefore, the development of a rapid, easy, and inexpensive method for melamine detection has become essential. With respect to its high sensitivity and reasonable instrument cost, fluorescence technique has become a favorite technique of detection. Since the strong H-bonding between cyanuric acid and melamine has been reported [8], we designed and synthesized new 1,8-naphthalimide fluorophores having a cyanuric acid pendant as a receptor for melamine. The synthesis and characterization of these new materials are reported herein. 2. Materials and Methods 2.1 Materials 1,8-Naphthalic anhydride, 4-bromo-1,8-naphthalic anhydride, ethanolamine, ethanol, cyanuric acid, potassium hydroxide (KOH), phosphorus tribromide (PBr3), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), magnesium sulphate (MgSO4), and 1,4dioxane were reagent grade and purchased from Sigma Aldrich. Organic solvents such as dichloromethane (CH2Cl2), hexane, and ethyl acetate (EtOAc) were commercial grade purchased from local suppliers and distilled prior to use. 2.2 Instrumentation The 1H NMR was measured by using 400 MHz 1H NMR spectrophotometer (Varian, USA) and reported as ppm in CDCl3 and DMSO-d6. The 13C NMR was measured on Bruker Mercury NMR spectrophotometer (Bruker, Germany) which were equipped at 100 MHz with chemical shifts reported as ppm in CDCl3 and DMSO-d6. A UV-2550 UV visible spectrophotometer (SHIMADZU,Japan) was used for the absorption studies. Emission spectra were acquired by a Carry Eclipse Fluorescence Spectrophotometer (Agilent Technologies). 2.3 Synthesis and Characterization 2.3.1. 2-(2-Hydroxyethyl)-1Hbenzo[d,e] isoquinoline 1,3(2H)-dione, (3) 1,8-Naphthalic anhydride (2.0 g, 10 mmol) and 3 mL ethanolamine were add into a round bottom flask, the mixture was heated 170 ºC under reflux conditions for 2 h. after the reaction was completed it was left to cool down to room temperature. The mixture was poured into 100 ml of cool water and the solid precipitate was filtered by vacuum filtration, and washed with cool water. After vacuum drying, compound 3 was obtained as brown solid in 91% yield; 1H NMR (400 MHz, CDCl3) δ (ppm): 8.64 (d, J = 7.3 Hz, 2H), 8.26 (d, J = 8.3 Hz, 2H), 7.79 (t, J = 7.8 Hz, 2H), 4.54 (m, 2H), 4.02 (t, J = 5.3 Hz, 2H). This Pure and Applied Chemistry International Conference 2015 (PACCON2015) 280 data was in good agreement with the literature report [9]. 2.3.2. 2-(2-Bromoethyl)-1H-benzo[d,e]isoquinoline1,3(2H)-dione (4) Compound 3 (0.15 g, 0.62 mmol) was dissolved in 20 mL of dichloromethane and then phosphorus tribromide (0.5 eq) was added drop wise. The reaction was stirred in an ice bath for 15 min. After the reaction was completed, it was diluted with water and extracted with CH2Cl2. The combined organic phase was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography using CH2Cl2 : Hexane (50:50) as the eluent to give 4 as white solid in 35% yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.62 (dd, J = 7.3 and 1.0 Hz, 2H), 8.23 (dd, J = 8.3 and 0.9 Hz, 2H), 7.77 (dd, J = 8.1 and 7.4 Hz, 2H), 4.61 (dd, J = 9.2 and 5.1 Hz, 2H), 3.67 (t, J = 7.2 Hz, 2H). 1-(2-(1,3-Dioxo-1H-benzo[de]isoquinolin-2 2.3.3 (3H)-yl)ethyl)-1,3,5-triazinane-2,4,6-trione (1) To a mixture of cyanuric acid (0.35 g, 2.7 mmol) and 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 0.08 ml, 0.5 mmol) in DMF (4 mL) at room temperature. The mixture was stirred at 100 ºC. The compound 3 (80 mg, 0.27 mmol) was added in the reaction for 24 h. The mixture was poured into water and extracted with EtOAc. The combined organic phase was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude substance was dissolved with DMSO and was poured into 100 ml of cool water, the white solid were collected by vacuum filtration and then dried overnight at room temperature in a vacuum oven to obtain 1 as white solid in 45% yield. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.35 (s, 2H), 8.49 (d, J = 7.2 Hz, 2H), 8.45 (d, J = 8.3 Hz, 2H), 7.86 (t, J = 7.8 Hz, 2H), 4.31 (brt, 2H), 4.06 (brt, 2H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 163.9, 150.0, 148.4, 134.4, 131.3, 130.8, 127.5, 127.2, 121.8, 39.1, 38.0. Elemental analysis : Calculated for C17H12N4O5 (MW 352.30) C 57.96, H 3.4, N 15.90%; found C 58.22, H 3.22, N 15.52%. 2.3.4. 6-Bromo-2-(2-hydroxyethyl)-1H-benzo [de]isoquinoline- 1,3(2H)-dione (5) The reaction of 4-bromo-1,8-naphthalic anhydride (5.01 g, 18.11 mmol) in 1,4 dioxane (25 mL) and 3 mL ethanolamine were added into a round bottom flask, the mixture was heated at 105 ºC under reflux conditions for 6 h. after cooling it down to room temperature. The mixture was poured into 100 mL of cool water and the solid precipitate was filtered by vacuum filtration and washed with cool water and then dried overnight at room temperature in a vacuum oven to give 5 as yellow solid in 95%yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.67 (d, J = 7.3 Hz, 1H), 8.59 (d, J = 8.5 Hz, 1H), 8.43 (d, J = 7.8 Hz, 1H), 8.05 (d, J = 7.8 Hz, 1H), 7.89 (m, 1H), 4.45 (t, J = 5.3 Hz, 2H), 3.99 (t, J = 5.3 Hz, 2H) [10]. 2.3.5. 6-Ethoxy-2-(2-hydroxyethyl)-1H- benzo[de]isoquinoline-1,3(2H)-dione (6) Compound 5 (1.01g, 3.16 mmol) was added into 30 mL of EtOH with KOH (0.17g, 3.1 mmol) and reflux for 4 h. Pour the mixture into 50 mL of water. And filtrate to collect the precipitant. The precipitant was washed with 30 mL of cool water and dried overnight at room temperature to afford 6 as dark yellow solid in 73%yield. 1H NMR (400 MHz, CDCl3) δ (ppm) : 8.61 (d, J = 7.9 Hz, 2H), 8.55 (d, J = 8.3 Hz, 1H), 7.71 (t, J = 7.8 Hz, 1H), 7.03 (d, J = 8.3 Hz, 1H), 4.48 (m, 2H), 4.36 (q, J = 7.0 Hz, 2H), 4.00 (m, 2H), 1.62 (t, J = 7.0 Hz, 3H). 2.3.6. 2-(2-Bromoethyl)-6-ethoxy-1H-benzo[d,e]isoquinoline-1,3(2H)-dione (7) Compound 6 (0.1 g, 0.36 mmol) was dissolved in 30 ml dichloromethane and then dropwise phosphorus tribromide (0.5 eq) was added drop wise. The reaction was stirred in an ice bath for 15 min. the reaction was completed, it was diluted with water and extracted with CH2Cl2. The combined organic phase was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography using EtOAc : Hexane (50:50) as the eluent to give 7 as pale yellow solid in 42%yield. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.60 (d, J = 7.7 Hz, 2H), 8.54 (d, J = 8.3 Hz, 1H), 7.70 (t, J = 7.8 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 4.58 (t, J = 7.2 Hz, 2H), 4.33 (q, J = 6.8 Hz, 2H), 3.64 (t, J = 7.2 Hz, 2H), 1.60 (t, J = 7.0 Hz, 3H). 2.3.7. 1-(2-(6-Ethoxy-1,3-dioxo-1H-benzo[d,e] isoquinolin-2(3H)-yl)ethyl)-1,3,5-triazinane-2,4,6trione (2) To a mixture of cyanuric acid (0.33 g, 2.6 mmol) and 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 2 eq.) in DMF (4 mL) at room temperature. The mixture was stirred at 100 ºC. The solid compound 6 (90 mg, 0.26 mmol) was added in the reaction. Reaction took place for 24h. After the reaction was completed, it was diluted with water and extracted with EtOAc. The combined organic phase was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude substance was dissolved with DMSO and was poured into 100 mL of cool water, the white solid was collected by vacuum filtration and then dried overnight at room temperature in a vacuum oven to provide 2 as pale yellow solid in 68%yield. 1 H NMR (400 MHz, DMSO-d6) δ (ppm): 11.37 (s, 2H), 8.55 (d, J = 8.3 Hz, 1H), 8.50 (d, J = 7.2 Hz, 1H), 8.44 (d, J = 8.3 Hz, 1H), 7.82 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 4.43 (m, 2H), 4.30 (brt, 2H), 4.05 (brt, 2H), 1.51 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 164.1, 163.4, 159.7, 149.9, 148.4, 133.4, 131.1, 128.8, 128.5, 126.3, 122.9, 121.7, 113.8, 106.9, 64.8, 37.8, 14.3. Elemental analysis : Calculated for C19H16N4O6 (MW 396.35) C 57.58, H 4.07, N 14.14%; found C 58.01, H 3.66, N 13.80%. Pure and Applied Chemistry International Conference 2015 (PACCON2015) 281 3. Results and discussion 3.1 Synthesis the compound 1 and 2 The synthesis of new target molecules were successfully conducted as shown in Scheme 1. The first reaction starting from commercially available 1,8naphthalic anhydride reacted with ethanolamine at 172oC to afford the 1,8-naphthalimide derivative 3 and then bromination reaction with phosphorus tribromide to provide 4 . The N-alkylation of 4 with cyanuric acid using DBU as a base in DMF gave fluorophore 1 in moderate yield. Fluorophore 2 was prepared via nucleophilic aromatic substitution of 4-bromo-1,8naphthalic anhydride with ethanolamine furnished 5, and nucleophilic replacement of bromine with an alkylether. The compound 2 is different from compound 1 in order to improve the quantum yield afforded 6. Next, 1,8-naphthalimide derivative 2 was prepared in good yield from 7 using the same condition described for compound 1. Figure 1. Normalized absorption and emission spectra of compound 1 and 2 in acetonitrile O HN Br OH O O O OH H2N N O O O N NH N O N O O O O PBr3 CH2Cl2 O 91% 35% 3 HN 4 O NH N H 1, 45% O DBU,DMF O HN OH O O O H2N N O OH N OH Br 95% 5 O O N N O O PBr3 CH2Cl2 KOH reflux,1,4-dioxane Br O O Br OH O O NH N O 73% O O O 42% 7 6 2, 68% Scheme 1. Synthesis of 1 and 2 3.2 Photo physical tests The absorption and emission spectra of each fluorophore were shown in Figure 2 and the related data were summarized in Table 1. The compounds exhibited maximum wavelength of absorption band at 332 nm of 1 and 366 nm of 2. An appearance of a longer wavelength, the maximum emission band of 2 at 430 nm, may cause from the effect of electron donating group (-OR), resulting in a bathochromic shift phenomenon [11]. And alkoxy was electron donating and electron withdrawing (imide) substituents which typically have charge-transfer lowest excited states. In addition, the quantum yield of the compound 2 is larger than that of the compound 1. Table 1: Photophysical properties of 1 and 2 in acetonitrile Cpd. Absorption Emission λabs (nm) ε (M-1 cm-1) λems (nm) Φ 1 332 5243 375 0.06 a 2 366 11,958 430 0.49b 2-Aminopyridine in 0.1 M H2SO4 (Φ = 0.60) and b Quinine sulphate in 0.1 M H2SO4 (Φ = 0.54) were used as references. a Pure and Applied Chemistry International Conference 2015 (PACCON2015) 282 4. Conclusions The compound 1 and 2 were successfully synthesized and characterized. Both of them have photo properties so they can be used for fluorescent sensor application. The compound 2 was found that the quantum yield more than the compound 1. It is dependent on the substituents at the 4-position. Acknowledgements This project is supported by the Ratchadapiseksomphot Endowment Fund of Chulalongkorn University (RES560530125-AM). References Suchý, P., Straková, E., Herzig, I., Staňa, J., Kalusová, R. and Pospíchalová, M., 2009, Interdisciplinary Toxicology, 2, 55. [2] Tyan, Y-C., Yang, M-H., Jong, S-B., Wang, C-K. and Shiea, J., 2009, Anal. Bioanal. Chem., 395, 729-735. [3] Liu, Y., Todd, E. D., Zhang, Q., Shi, J.-R. and Liu, X.J., 2012, J. Zhejiang Univ. Sci. B., 13, 525-532. [4] Li, J., Yang Qi, H. and Ping Shi, Yan., J., 2009, J. Chromatogr. A., 1216, 5467-5471. [5] Gao, L. and Jönsson, J. Å., 2012, Analytical Letters, 45, 2310-2323. [6] Goscinny, S., Hanot, V., Halbardier, J. F., Michelet, J. Y. and van Loco, J., 2011, Food Control., 22, 226-230. [7] Garber, E. A. E., 2008, J. Food Prot., 71, 590-594. [8] Sanji, T., Nakamura, M., Kawamata, S., Tanaka, M., Itagaki, S. and Gunji, T., 2012, Chem. Eur. J.,18, 15254 - 15257 [9] Wan, X., Liu, T. and Liu, S., 2011, Langmuir 27, 4082-4090. [10] Li, H.-T., Jiang, Z.-Q., Zheng, J., Wang, X., Pan, Y., Wang, F. and Yu, S.-Q., 2006, Res. Chem. Intermediat., 32, 43-57. [11] Georgiev, N.I. and Bojinov, B.V., 2012, J. Lumines., 132, 2235-2241. [1] Pure and Applied Chemistry International Conference 2015 (PACCON2015)





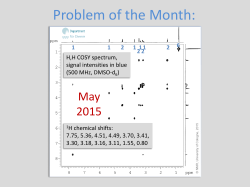




© Copyright 2025