
fulltext
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine Modeling glioblastoma heterogeneity to decipher its biology YUAN XIE ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2016 ISSN 1651-6206 urn:nbn:se:uu:diva-278529 Dissertation presented at Uppsala University to be publicly examined in Rudbecksalen, Dag Hammarskjold v 20, Uppsala, Friday, 15 April 2016 at 09:15 for the degree of Doctor of Philosophy (Faculty of Medicine). The examination will be conducted in English. Faculty examiner: PhD, head of the Seve Ballesteros Foundation Brain Tumor group Massimo Squatrito (BBVA Foundation – CNIO Cancer Cell Biology Programme). Abstract Xie, Y. 2016. Modeling glioblastoma heterogeneity to decipher its biology. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 49 pp. Uppsala: Acta Universitatis Upsaliensis. Glioblastoma multiforme (GBM) is the most common and lethal form of primary brain tumor that mainly affects adults. GBM displays remarkable intra- and inter-tumoral heterogeneity and contains a subpopulation of cells named glioma stem cells that is believed to be responsible for tumor maintenance, progression and recurrence. We have established and characterized a biobank of 48 cell lines derived from GBM patients. The cells were explanted and maintained as adherent cultures in serum-free, defined neural stem cell medium. These GBM cells (GCs) displayed NSC marker expression in vitro, had orthotopic tumor initiating capability in vivo, harboured genomic alterations characteristic of GBM and represented all four TCGA molecular subtypes. Our newly established biobank is also connected with a database (www.hgcc.se) that provides all molecular and clinical data. This resource provides a valuable platform of valid in vitro and in vivo models for basic GBM research and drug discovery. By using RCAS/tv-a mouse models for glioma, we found that GBMs originating from a putative NSC origin caused more tumorigenic GCs that had higher self-renewal abilities than those originating from putative glial precursor cell origin. By transcriptome analysis a mouse cell origin (MCO) gene signature was generated to cluster human GCs and GBM tissue samples and a functional relationship between the differentiation state of the initially transformed cell and the phenotype of GCs was discovered, which provides the basis for a new predictive MCObased patient classification. LGR5 was found to be highly expressed in the most malignant mouse GC lines of putative NSC origin and also enriched in proneural GBMs characterized by PDGFRA alterations and OLIG2 up-regulation. By overexpressing or depleting LGR5 we discovered that high LGR5 expression in proneural GC lines increased the tumorigenicity, self-renewal and invasive capacities of the cells and could potentiate WNT signalling through its ligand RSPO1. Through transcriptome analysis we identified the candidate genes CCND2, PDGFRA, OLIG2, DKK1 that were found to be regulated by LGR5. In the last study, we found that mouse OPCs could initiate both astrocytic and oligdendroglial gliomas, which indicated that oncogenic signalling is dominant to cell of origin in affecting the histology of gliomas. Keywords: Glioblastoma multiforme, biobank, GBM cells, cell of origin, LGR5, OPCs Yuan Xie, Department of Immunology, Genetics and Pathology, Rudbecklaboratoriet, Uppsala University, SE-751 85 Uppsala, Sweden. © Yuan Xie 2016 ISSN 1651-6206 urn:nbn:se:uu:diva-278529 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-278529) To my family ”A will finds a way” -Orison Swett Marden List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I Xie, Y.*, Bergström, T.*, Jiang, Y.*, Johansson, P.*, Marinescu, V. D., Lindberg, N., Segerman, A., Wicher, G., Niklasson, M., Baskaran, S., Sreedharan, S., Everlien, I., Kastemar, M., Hermansson, A., Elfineh, L., Libard, S., Holland, E.C., Hesselager, G., Alafuzoff, I., Westermark, B.#, Nelander, S.#, Forsberg-Nilsson, K.#, and Uhrbom, L.# (2015). The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes. EBioMedicine, 2 (10):1351-1363. II Jiang, Y.*, Marinescu, V.D.*, Xie, Y., Jarvius, M., Haglund, C., Pendekanti, P., Olofsson, S., Lindberg, N., Olofsson, T., Hesselager, G., Alafuzoff, I., Fryknäs, M., Larsson, R., Nelander, S.#, and Uhrbom, L.# (2016). Malignancy and drug sensitivity of glioblastoma cells are affected by the cell of origin. Submitted. III Xie, Y., Sundström, A., Marinescu, V.D., Tan, E-J., Pendekanti, P., Tirfing, M., Blokzijl, A., Jin, C., Chen, L., Essand M., Swartling, F.J., Nelander S., Jiang Y., and Uhrbom L. (2016). High LGR5 expression in proneural glioblastoma cells contributes to increased self-renewal and invasiveness. Manuscript. IV Lindberg, N., Jiang, Y.*, Xie, Y.*, Bolouri, H., Kastemar, M., Olofsson, T., Holland, E.C., and Uhrbom, L. (2014). Oncogenic signaling is dominant to cell of origin and dictates astrocytic or oligodendroglial tumor development from oligodendrocyte precursor cells. The Journal of Neuroscience, 34 (44): 14644-14651. *, # These authors contributed equally to this work. Reprints were made with permission from the respective publishers. Other papers by the author: • Segerman, A., Niklasson, M., Haglund C.*, Bergström, T.*, Jarvius, M.*, Xie, Y., Caglayan, D., Westermark, A., Hermansson, A., Kastemar, M., Ali, Z.N., Berglund, M., Sundström, M., Hesselager, G., Uhrbom, L., Gustafsson, M., Larsson, R., Fryknäs, M., Segerman, B.#, and Westermark, B.# (2016). Glioblastoma exhibits phenotypic microheterogeneity and harbors pre-existing multiresistant clones with a mesenchymal transition signature. Submitted. • Sreedharan, S.*, Xie, Y.*, Pendekanti, P.*, Sundström, A., Jarvius, M., Libard, S., Fryknäs, M., Larsson, R., Alafuzoff, I., Swartling, F.J., and Uhrbom L. (2016). Molecular and phenotypic influences of cell of origin on pediatric high-grade glioma. Manuscript. *, # These authors contributed equally to this work. Contents Classification of glioma ................................................................................ 11 Histological classification......................................................................... 11 Transcriptome-based molecular classification ......................................... 12 Epigenetic-based molecular classification................................................ 14 Core pathways in human GBM ..................................................................... 15 RTK signalling pathways ......................................................................... 15 p53 and RB pathways ............................................................................... 18 Mouse models of glioma ............................................................................... 19 Chemical mutagen-induced models.......................................................... 19 Xeno- or allograft transplantation models ................................................ 19 Germline modification models ................................................................. 20 Somatic cell genetic modification models ................................................ 20 RCAS/tv-a glioma model ..................................................................... 21 Cellular origin of glioma ............................................................................... 23 Neural stem cells ...................................................................................... 23 Glial precursor cells .................................................................................. 23 Oligodendrocyte precursor cells ............................................................... 24 Sources of Heterogeneity within Cancer ....................................................... 25 Intrinsic Genetic/Epigenetic Clonal Evolution ......................................... 25 Extrinsic Environmental Effects ............................................................... 25 Cancer Stem Cell Model........................................................................... 26 Glioma Stem Cells ............................................................................... 26 Combined Model ...................................................................................... 27 LGR5 ............................................................................................................. 28 LGR5 and Wnt signalling ......................................................................... 28 LGR5 in cancer ......................................................................................... 29 LGR5 in GBM .......................................................................................... 30 Present investigations .................................................................................... 31 Paper I. ...................................................................................................... 31 The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes .................................... 31 Paper II. .................................................................................................... 32 Malignancy and drug sensitivity of glioblastoma cells are affected by the cell of origin .............................................................................. 32 Paper III. ................................................................................................... 34 High LGR5 expression in proneural glioblastoma cells contributes to increased self-renewal and invasiveness.......................................... 34 Paper IV. ................................................................................................... 36 Oncogenic signalling is dominant to cell of origin and dictates astrocytic or oligodendroglial tumor development from oligodendrocyte precursor cells ........................................................... 36 Future perspective ......................................................................................... 38 Acknowledgements ....................................................................................... 40 References ..................................................................................................... 43 Abbreviations ALV APC APCs ARF BMP4 BTSC CDKN2 CNAs CNP CNS COO CRC CSC eGFP EGF EGFR eNOS FGF G-CIMP GBM GC GEF GFAP GPR GSC HGCC HMG IDH1 INK LGR5 LOH avian leucosis virus adenomatous polyposis coli astrocytic precursor cells p14/p19 Arf bone morphogenetic protein 4 brain tumor stem cell cyclin-dependent kinase inhibitor 2A copy-number aberrations 2’, 3’-cyclic nucleotide 3’-phosphodiesterase central nervous system cell of origin colorectal cancer cancer stem cell enhanced green fluorescent protein epidermal growth factor epidermal growth factor receptor endothelial nitric oxide synthase fibroblast growth factor glioma-CpG island methylator phenotype glioblastoma multiforme GBM cells guanine exchange factor glial fibrillary acidic protein G-protein coupled receptor glioma stem cell Human Glioblastoma Cell Culture high mobility group isocitrate dehydrogenase 1 inhibitor of CDK4 leucine-rich repeat-containing G-protein coupled receptor 5 loss of heterozygosity LRP MAPK MADM MCO MDM2 Mes MLL5 MMLV NF1 NO NSCs O2A Olig2 OPCs PDGF PDGFR PDGFRA PI3K PIP2 PN Prolif PTEN PVN RB RCAS RSPO RTK SVZ TCGA TMA TP53 tv-a VEGF WHO low density-lipoprotein receptor related protein mitogen-activated protein kinase mosaic analysis with double markers mouse cell origin murine double minute 2 mesenchymal mixed lineage leukemia 5 moloney murine leukemia virus neurofibromin 1 nitric oxide neural stem cells oligodendrocyte/ type-2 astrocyte oligodendrocyte transcription factor oligodendrocyte progenitor/precursor cells platelet-derived growth factor platelet-derived growth factor receptor platelet-derived growth factor receptor alpha phosphoinositide 3-kinase phosphatidylinositol-4, 5-biosphosphate proneural proliferative phosphatase and tensin homolog perivascular niche retinoblastoma 1 replication competent ALV splice acceptor R-Spondin proteins receptor tyrosine kinase subventricular zone The Cancer Genome Atlas Research Work tissue microarray tumor suppressor protein 53 receptor for subgroup A avian sarcoma and leukosis virus vascular endothelial growth factor World Health Organization Classification of glioma Malignant gliomas are the most common and aggressive primary intracranial brain tumor mainly affecting adults. The most frequent and malignant glioma is called glioblastoma multiforme (GBM). Approximately 5% of the GBM patients will survive five years after diagnosis despite implementation of intensive therapeutic strategies and medical care1. The incidence of GBM is much higher in older people with the highest in 75 to 84 years old patients1. Due to its features of diffuse infiltration, resistance to radio/chemotherapy, and remarkable intra- and inter-tumoral heterogeneity, the prognosis is very poor and currently there is no cure for GBM patients2. Histological classification Gliomas are categorized morphologically as ependymomas, astrocytomas and oligodendrogliomas. Tumors are further histologically classified into four (I-IV) grades according to the World Health Organization (WHO) malignancy scale3. Grade I tumors are biological benign with a low proliferation index, and can often be cured by surgical resection alone. These gliomas usually occur in children or young adults. Grade II tumors begin to infiltrate into surrounding brain tissues, but with a relatively low proliferation potential. Several grade II tumors tend to transform to higher grades of malignancy. For instance, diffuse astrocytomas with a low-grade can progress to highgrade anaplastic astrocytomas and GBMs. Grade III tumors exhibit nuclear atypia, increased anaplasia and mitotic activity. In most cases, patients with grade III tumors are treated by radio/chemotherapy. Grade IV tumors are the most malignant and most frequent and account for 45.6% of all gliomas1. These tumors are also termed GBM and often display pseudopalisading necrosis and extensive vascular proliferation, considered to be hallmarks of GBM. Approximately 74,000 GBMs are diagnosed worldwide per year4. The median survival for GBM patients ranges from 12 to 15 months despite intensive treatment5. GBMs are further subdivided into primary and secondary GBM subtypes. Primary GBMs account for approximately 90% of all GBMs, and usually occur in older patients, while secondary GBMs typically occur in younger patients (< 45 years). Primary GBMs are diagnosed de novo while secondary GBMs are derived from a prior diagnosis of a lower grade glioma. Primary and second11 ary GBMs have indistinguishable histopathological, but their genomes and transcriptomes are remarkably different. Figure 1. Histological classification of astrocytic tumors based on WHO malignancy scale. Modified from1,3,5-7. Transcriptome-based molecular classification Human malignant gliomas exhibit a great level of inter- and intra-tumoral molecular heterogeneity that is histopathologically indistinguishable. Based on large-scale analysis of genomes, transcriptomes, and proteomes of tumor samples, the first molecular classification of high-grade gliomas (HGGs = grade III-IV gliomas) displayed three molecular subtypes: proneural (PN), proliferative (Prolif), and mesenchymal (Mes)8. These subtypes displayed a association with histological tumor grade. PN included almost all the WHO grade III tumors and a part of grade IV tumors. In contrast, Prolif and Mes are exclusively grade IV tumor with necrosis. Patients classified into PN were around 40 years old with relatively longer survival. However, patients from Prolif and Mes were about 50 years old with shorter survival. Loss of PTEN and EGFR amplification could be found in both Prolif and Mes subtypes, but not in PN. 12 Subsequently, The Cancer Genome Atlas research network (TCGA) provided a robust gene-expression based molecular classification of adult GBMs that is currently widely used. In this classification, GBMs are divided into four molecular subtypes: classical, mesenchymal, neural, and proneural9. The classical group harbours high-level EGFR amplification, homozygous CDKN2A deletions, and a distinct lack of TP53 mutations. Chromosome 7 gain paired with chromosome 10 loss is also one main feature in classical samples. The mesenchymal subtype is characterized with predominantly hemizygous NF1 deletion and lower level of NF1 mRNA expression. This subtype also exhibited expression of mesenchymal markers (CHI3L1/YKL40 and MET) and astrocytic markers (CD44 and MERTK). The neural samples have no defining mutations and an expression of markers that is quite similar to normal brain. The proneural class harbours PDGFRA alterations or point mutation in IDH1, and frequent TP53 mutations. This group displayed high expression of oligodendrocytic lineage genes, such as PDGFRA and OLIG2. Moreover, a subset of the proneural group was found display global hypermethylation, a feature termed glioma-CpG island methylator phenotype (G-CIMP), often overlapping with the IDH1 mutated samples, and these patients were younger at diagnosis and had improved survival10,11. Accumulating evidence also indicate the remarkable intratumor heterogeneity of GBM which can be an underlying cause for the treatment failure. Different samples from the same GBM patient can be classified as different TCGA subtypes12. Individual cells13 or single cell-derived clones14 from the same GBM patients can also be grouped as different subtypes by transcriptome analysis. Table 1. Molecular classification of GBM based on large-scale analysis of genomes and transcriptomes by TCGA. 13 Epigenetic-based molecular classification Furthermore, an epigenetic-based GBM classification that also included pediatric GBMs has revealed six distinct DNA methylation clusters15. Hotspot H3F3A mutations affecting two amino acids (K27 and G34) have been identified almost exclusively in GBMs from children, that were mutually exclusive with IDH1 mutations. Based on correlation with mutations, gene expression profile and DNA copy-number aberrations (CNAs), six distinct DNA methylation clusters are identified, “IDH”, “K27”, “G34”, “RTK I (PDGFRA)”, “mesenchymal” and “RTK II (Classic)”. “IDH1” group contains a majority of IDH1 mutated tumors and exhibits concerted hypermethylation, which is clearly mapped to G-CIMP+ cluster10. “proneural” signature is found in all the tumors from “IDH1” cluster9. “K27” group is also clearly enriched in “proneural” expression. Nevertheless, for ”G34” cluster, it displays widespread hypomethylation especially in nonpromoter regions. In all the three clusters of IDH1, K27 and G34, frequent TP53 mutations and lack of detected CNAs (amplification of EGFR, deletion of CDKN2A, chromosome 7 gain paired with chromosome 10 loss) were observed. In addition, “RTK I (PDGFRA)”, “mesenchymal” and “RTK II (classical) clusters are respectively highly enriched for proneural, mesenchymal, and classical gene expression signatures described by TCGA9. It is known that dysregulation of H3.3 is through mutations in pediatric GBM. Adult GBMs also display the dysregulation of H3.3 but through epigenetic repression. It has been revealed that mixed lineage leukemia 5 (MLL5) and H3.3 together play a role in maintaining cell self-renewal, a key character of cancer cell contributing to GBM recurrence16. 14 Core pathways in human GBM Multi-dimensional molecular analysis has identified three core pathways in human GBM pathogenesis, including RTK signalling, and the p53 and RB tumor suppressor pathways. TCGA has found 74% of 206 human GBM samples carried aberrations in all three pathways, suggesting that cooperation among three pathways is a core requirement in controlling glioma cell proliferation, apoptosis, necrosis, angiogenesis and metastasis17. RTK signalling pathways Based on the latest study of GBM by TCGA, genetic aberrations have been found in up to 90% of the RTK/PI3K/RAS pathway11. Receptor tyrosine kinase (RTK) signalling pathways are critical targets for glioma initiation and progression. Several mechanisms are involved in activating the RTK pathways, such as receptor amplification, abnormal activity through autocrine stimulation or receptor mutations causing constitutive activity. In nomal cells RTKs are activated upon ligand binding (e.g. EGFs, FGFs, PDGFs or VEGFs) to their specific RTK that induces dimerization of RTKs and autophosphorylation of intracellular domains. The phosphorylated RTKs then activate corresponding downstream effector pathways, including Ras/MAPK, PI3-K, PLC-γ, and JAK-STAT, which can regulate cellular responses such as proliferation, survival and migration18. 15 Ligand (EGF,FGF,VEGF or PDGF) Dimerization RTK RTK Cell membrane RAS NF-1 Raf SOS Grb-2 p p p p PI3K MEK PIP2 PIP3 PIP2 Akt PTEN ERK Proliferation Survival Cell cycle progression Migration Nuclear Membrane CDKN2A Mutant Arf p16INK4a p14/p19ARF S G2 MDM2 MDM2 Cell cycle restriction Arf p53 M G1 p p53 p p Proteasome p21 p pRb p p E2F pRb p p CDK4 Cyclin CDK6 Figure 2. Major signalling pathways in human GBM. Modified from19. Red prints indicate oncogenes, and green prints indicate tumor suppressor genes. 16 PDGFR signalling is an essential regulator of gliomagenesis. There are four PDGF ligands PDGF-A, -B, -C, -D, that form of homo- or heterodimers, which may bind to the receptors PDGFR-αα, PDGFR-αβ or PDGF-ββ20. PDGF is secreted by type-1 astrocytes and promotes the division and development of bipotential oligodendrocyte/ type-2 astrocyte (O2A) progenitor cells21-23. In the normal brain PDGFR signalling is considered to play a predominant role in oligodendrocyte development22. It has been found that PDGFR-α is downregulated in glial progenitors that differentiate into oligodendrocytes, and PDGF-AA deficient mice have a great decrease in the number of glial progenitors and oligodendrocytes24. In the adult mouse brain, PDGFR-α is restricted to the ventricular and subventricular zones (SVZ) of the lateral ventricles, which is deemed as the biggest niche of adult neurogenesis and the most likely source of gliomas25. PDGFRα+ cells have also been found scattered throughout the white matter and cerebral cortex of adult rodents26. An important role for PDGFR signalling in human GBM was implied for the first time several decades ago when coexpression PDGF ligands and receptors were described in glioma cells27, that was shown to result in increased cell proliferation due to an autocrine loop2830 . Unsupervised analysis of 251 GBMs, PDGFRA was found altered in 13% of all the samples. The majority of alterations in GBM involve EGFR signalling11,17. EGF mRNA expression could be detected in all regions examined including cerebellum, cerebral cortex, brain stem, olfactory bulb, hippocampus, basal hypothalamus, striatum and thalamus during central nervous system (CNS) development in rat31. On embryonic day 14, EGF expression could be detected and continued to be detected to the postnatal period in the whole brain31. Cellular localization and finding of EGFR support the notion of the roles of EGF in CNS development. EGFR is also found to be expressed in various region of the CNS32. In primary GBM, EGFR is frequently activated. The most common EGFR VIII mutant is the deletion of extracellular domain (exon 2-7), which result in a constitutively active receptor33,34. According to the latest study by TCGA, up to 57% of the GBMs displayed EGFR alterations, including mutation, rearrangement, altered splicing, and /or focal amplification11. Ras/MAPK and PI3-K are two major effector pathways activated by RTKs. Ras are part of the monomeric GTPase superfamily of proteins. Ras are located in the inner surface of the plasma membrane and transmit extracellular signals to promote the activities of cells, such as cell growth, proliferation and differentiation. Autophosphorylation of intracellular receptor domains allows guanine exchange factor (GEF) to attach to the receptor by the adaptor proteins SOS and Grb-2. GEF induces the exchange of GDP to GTP, which result in activation of Ras. Furthermore, Ras-GTP can be inactivated by Ras-GAP proteins (e.g. NF1). NF1 is a tumor suppressor in glioma, inactivating mutations or homozygous deletions of NF1 can be found in 10% 17 of GBMs11. The major downstream target of Ras-GTP is mitogen-activated protein kinase (MAPK), which is involved in the regulation of cell proliferation. The other important effector of RTK activation, PI3K (phosphinositide 3kinase), acts by phosphorylating PIP2 to PIP3 that in turn activates its downstream target Akt, which plays a vital role in cell survival and cell proliferation. Akt mutations are very rare but the high activity has been seen in most GBM samples17. However, tumor suppressor protein PTEN catalyses dephosphorylation of PIP3 to PIP2 and counteracts the PI3K activity. Mutations or homozygous deletions of PTEN can be detected in 41% of GBMs11. p53 and RB pathways According to TCGA, at least one genetic alteration has been identified in 85% and 79% of p53 and RB pathways respectively11, that is most GBMs have mutations of both these pathways. The essential components of the RB pathway are CDKN2A, CDK4/6 and RB and in the p53 pathway CDKN2A, MDM2/4 and p53. The CDKN2A locus is thus shared by both p53 and RB signalling and encodes two tumor suppressor proteins, p16INK4a and p14ARF. p16INK4a inhibits CDK4 and CDK6 that promote activation of RB. Activation of Rb proteins bind E2F family proteins, which in turn inhibit the cell cycle to move on to the S phase. The other tumor suppressor encoded by CDKN2A, p14ARF, can bind to and inactivate MDM2, an E3 ubiquitin ligase that cause degradation of p53. Furthermore, p53 can bind to promoters of many downstream genes that regulate cell cycle arrest, DNA repair and programmed cell death. 18 Mouse models of glioma Mouse models are the favorite animal models to study human cancers due to their genomic similarity and short generation period. Mouse models can be used to study complex tumor characteristics including malignancy, invasion, and metastasis, which cannot be modeled in cell culture systems. Moreover, mice are essential tools as preclinical models for identifying potential therapeutic targets for tumorigenesis. To date, there are four predominant strategies employed that mimic human gliomagenesis: chemical mutagen-induced models, xeno- or allograft transplantation models, germline genetic modification models and somatic cell genetic modification models35. Chemical mutagen-induced models DNA alkylating agents are commonly used to generate point mutations to induce glioma36-38. Although the histology of induced tumors show similarity to equivalent tumors in human, the initiating mutation responsible for tumor induction and the cell of origin are not known in this model. Xeno- or allograft transplantation models This strategy has been widely used in preclinical trials39,40 and to examine the tumor initiating capacity of brain tumor stem cells (BTSCs). Human or mouse glioma cell lines are injected subcutaneously or orthotopically into immunocompromised mice. These cells usually grow fast and tumor incidence is high. These models have several good characteristics including defined and reproducible tumor formation and time to death41. However, genetic alterations and immunological interaction between tumor and host are not known in these models, which is not ideal for investigating early stages of tumor development but good tools for drug test and preclinical studies. 19 Germline modification models The formation of glioma is a multistep process that involve altered growthfactor signalling and altered cell-cycle arrest. More genetically accurate rodent model is developed by gain of function transgenic approaches and loss of function targeted deletion strategies. By applying the promoter that drives a construct’s expression, knock-in mice can express a gene of interest, or called oncogene in all the cells. On the contrary, transgenic mice can lose expression of tumor suppressor genes. By applying the Cre/Lox technology, inducible conditional knock-out or knock-in models can be made through recombining a pair of target DNA sequences (named lox sequence) by Cre recombinase42. This technology is widely used in germline modification models. However generation of germline modification models is a timeconsuming process. Somatic cell genetic modification models By injecting retroviral vectors, genes of interest are delivered into cells postnatally. Unlike germline modification, somatic cell genetic modifications are not inheritable. To date, two common systems are available including MMLV (moloney murine leukemia virus) and ALV (avian leucosis virus) single–stranded RNA virus based approaches43. MMLV has the ability to infect any dividing cell types. Obviously, the cell of origin of gliomagenesis cannot be specified by using this system. However, ALV has a limited infection capacity restricted to avian cells because their expression of the tv-a receptor. Mammalian cells can be made susceptible to ALV infection through transgenic expression of tv-a. RCAS (replication competent ALV splice acceptor) is an ALV retrovirus that is used as vehicle to transfer genes of interest into transgenic mice that express tv-a, the avian cell surface receptor for RCAS. Nowadays, the RCAS/tv-a system is widely used in transferring oncogenes into selected somatic cell types, which may allow for spatiotemporal control of tumorgenesis. It is an excellent model for addressing the cell of origin of glioma43. Recently, an efficient and promising targeting method CRISPR/Cas9 has emerged to model gliomagenesis44. By applying this technology to delete multiple genes (Trp53, Pten, Nf1) in the mouse brain, GBM-like tumor will develop45. 20 RCAS/tv-a glioma model To obtain cell type-specific gene transfer by RCAS tv-a transgenic mice are needed. Figure 3. The RCAS/tv-a mouse model. Three different transgenic tv-a mouse lines N/tv-a46, G/tv-a47 and C/tv-a48 were used in our glioma studies. There are many tv-a transgenic mouse lines available46-52, but in our glioma studies we have used three different tv-a transgenic mouse lines, Nestin/tv-a, GFAP/tv-a and CNP/tv-a. The Nestin promoter is used to control the expression of tv-a in N/tv-a mice, which direct infection to CNS stem and progenitor cells47. In G/tv-a mice the glial fibrillary acidic protein (GFAP) promoter is used that direct infection to mature astrocytes and NSCs46, and in the C/tv-a mice recently established in our lab, the CNP (2',3'-cyclic nucleotide 3'-phosphodiesterase) promoter is used to drive infection in late oligodendrocyte progenitor cells (OPCs) and mature oligodendrocytes48. All 21 tv-a mouse lines have been crossed with mice deficient for the tumor suppressor genes of Cdkn2a locus, Ink4a, Arf or combined loss of both. With these mouse lines we and others have modeled many types and grades of gliomas. For instance, with RCAS-PDGF-B oligodendrogliomalike tumors could be induced in all three tv-a lines48,53-55. Combined K-ras and Akt infection generated malignant gliomas in N/tv-a but not G/tv-a mice55. The additional loss of Ink4a or Arf in both N/tv-a and G/tv-a mice lead to higher tumor incidence and malignancy55,56. 22 Cellular origin of glioma The cell of origin in human GBM is unknown. Investigations from mouse models imply that many cell types of the CNS can be the origin. Development of cells in CNS occurs in a hierarchical way, from NSCs to committed precursor cells that in turn differentiate into neurons, ependymal cells, astrocytes and oligodendrocytes. Based on results from animal models many of these cell types have the potential to serve as a cellular origin of glioma. Neural stem cells NSCs mainly reside in the SVZ and hippocampus. Human brain tumors are frequently located in this germinal region, SVZ57. By evaluation of MRI scans in 100 glioma patients, up to 93% cases the lesion connected to SVZ58. NSCs have self-renewal capacity, and can differentiate into neurons and macroglia (astrocytes and oligodendrocytes). Several surface markers are available to characterize both normal NSCs and GSCs, like Sox259, Nestin60, CD13361, GFAP57, and GFAP delta57,62. Nevertheless, none of them is universal stem cell marker. Combination of p53 loss and activation of Ras signalling via Nf1 inactivation is demonstrated to initiate malignant astrocytoma with 100% penetrance. Histopathology analysis of all these tumors reveals that all the early pre-symptomatic lesions reside within SVZ of lateral ventricle, indicating that NSCs in SVZ region may serve as a cell-of-origin63. Inactivation of tumor suppresors p53, Nf1 and Pten in neural stem cells is certified to be necessary and sufficient to develop malignant astrocytoma64. Putatively NSC derived glioma stem cells are demonstrated to sustain GBM growth and recurrence after chemotherapy65. Glial precursor cells GFAP is expressed in both radial glia cells66 and NSCs of the SVZ67, which has been widely used as a promoter to control many different oncogenic mutations. Somatic delivery of PDGFB into GFAP+ cells results in oligodendrogliomas or oligo-astrocytomas in 40% mice by applying RCAS/tv-a mouse model, and loss of Ink4a-Arf shortens latency and increases malignancy of glioma53. Inactivation of pRb, p107 and p130 in GFAP+ cells in23 duced high-grade astrocytoma, and the tumor development was accelerated by heterozygous deletion of PTEN but not P5368. Lentiviral transduction of shNF1-shp53 in GFAP+ cells can give rise to malignant glioma69. Oligodendrocyte precursor cells CNP is a highly specific marker in the CNS for late development of OPCs or in mature oligodendrocytes. It is applied as a promoter to control oncogenic mutations in C/tv-a mice that is developed in our lab48. RCAS-PDGFB transfer to CNP+ cells can induce WHO grade II glioma with a penetrance of 33%. Although infected cells are CNP+, the tumor cells start to express earlier OPCs markers, like SOX2, SOX10, OLIG2, NG2, and PDGFR, indicating a process of dedifferentiation occur in target OPCs. NG2+ oligodendroglioma cells show sensitivity to chemotherapy and have limited self-renewal capacity, which indicate a progenitor origin for these cells70. Additionally, a new mouse model “MADM” (mosaic analysis with double markers) allows to trace entire tumorigenic process when initial mutation p53/Nf1 sporadically in NSCs. Interestingly, aberrant proliferation prior to tumor lesion can only be discovered in OPCs. Transcriptome analysis of these tumors also reveals obvious OPC feature71. Recently, two phenotypically and molecularly distinct subtypes of GBM has been discovered, which arise from functionally different CNS progenitors. This finding point to that the cell of origin plays an essential role in determining GBM subtype diversity72. 24 Sources of Heterogeneity within Cancer Intrinsic Genetic/Epigenetic Clonal Evolution The concept of genetic clonal evolution was presented in 1970. It suggests that a tumor is initiated from one mutated single cell that acquires a selective growth advantage over the adjacent cells. After a latency period, neoplastic proliferation eventually leads to genetic instability in the expansion of a tumor cell population73. Also, a central role of epigenetic processes in cancer development have been frequently studied in the past decades74. Upon intrinsic genetic/epigenetic clonal evolution, heterogeneity arises within tumors in a stochastic manner and through a selection process. Extrinsic Environmental Effects Heterogeneity can also arise in response to extrinsic environmental factors. Cancer cells adjacent to the perivascular niche (PVN) show differences from the cells located further away from the PVN. One study shows that endothelial nitric oxide synthase (eNOS) is significantly increased in tumor vascular endothelium that is adjacent to glioma cells. Only the perivascular glioma cells show expression of Nestin, Notch and the nitric oxide (NO) receptor, but not the cells further from blood vessels75. This indicates that only a population of glioma cells adjacent to blood vessels can respond to NO signalling. 25 Figure 4. Sources of heterogeneity within cancer. Modified from76. Cancer Stem Cell Model The cancer stem cell (CSC) model suggests that tumors are mainly initiated by a rare fraction of self-renewing, multipotent cells with stem cell capabilities77-79. There is now compelling evidence that various cancers, both leukemias and solid tumors are organized in a hierarchical manner and sustained by the so-called CSCs (or tumor initiation cells) which can generate full repertoire of tumor cells80. Glioma Stem Cells GSCs are a subpopulation of cells suggested to be responsible for glioma maintenance, therapeutic resistance and recurrence. To date, three functional criteria are used to characterize GSCs: (1) They are tumorigenic after intracranial orthotropic transplantation into NOD-SCID mice. (2) GSCs can give rise to both tumorigenic and non-tumorigenic cells, establishing a cellular hierarchy. (3) They have increased self-renewal capacity and multi-lineage differentiation potentials81. As GSCs can be a potential therapeutic target, there is a great interest in finding a reliable cell-surface marker to identify and isolate them. CD133 (prominin-1) was the first marker used to isolate GSCs. These CD133+ cells held the capacity to proliferate and self-renewal in vitro and lacked expression of neural differentiation markers. As few as 100 CD133+ were proved to be sufficient for initiation of human brain tumors in NOD-SCID mice, while as much as 50,000 to 100,000 CD133- cells could not form tumors in the same mice. It suggested that CD133+ human brain tumor cell fraction contain GSCs that exclusively induced tumor formation82. Furthermore, CD133+ 26 cells exhibit the ability to activate DNA damage checkpoint in response to radiation, which is deemed as the source of tumor recurrence after radiation83. However, increasing controversy argues that CD133- cells also have the capability to initiate tumor in nude mice and give rise to CD133+ cells84. Approximately 40% of freshly isolated GBM biopsies do not contain CD133+ cells. It seems that the ability of self-renewing and proliferation in human brain tumor might not be restricted to an exclusive subset of CD133+ stem-like cells. Over the last decade, several additional markers have emerged to enrich GSCs, like CD4485, L1CAM86, A2B587,88, SSEA-181 and integrin alpha 689. Some GSCs markers are also embryonic stem cell marker for normal brain development, like Sox290-92, Nestin93,94, Nanog91 and Oct491,95,96. As GBM displays remarkable intra- and intertumoral heterogeneity, it seems unlikely to discover a universal marker for GSCs. Instead, trying to identify GSC markers coupled to subgroups of GBMs, such as the molecular subtype or as yet novel subgroups, will probably be a more successful strategy. Combined Model The above three sources of heterogeneity are not mutually exclusive and may apply to variable extents depending on the type of cancer. Cancer that depend on the CSC model could also follow genetic/epigenetic clone evolution and in response to extrinsic environmental effects. In addition, since many cell types in the mouse can develop glioma it is not unrealistic to hypothesize that so is the case also in human glioma. In line with such reasoning it may also be hypothesized that different cellular origin may contribute to the inter-tumoral heterogeneity found in GBM. 27 LGR5 Leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5) is also known as G-protein coupled receptor 49 (GPR49) or G-protein coupled receptor 67 (GPR67). It is a 7-transmembrane receptor that has gained prominence as a stem cell marker in the intestine, stomach and hair follicle, however its expression can be found across a diverse range of tissues such as muscle, placenta, spinal cord and brain. LGR5 and Wnt signalling LGR5 is a member of wnt signalling pathway that controls stem cell selfrenewal and differentiation. Wnt receptor complex that activates canonical pathway contains two components: a member of the Frizzled family and a member of the low density-lipoprotein receptor related protein (LRP5 or LRP6). R-Spondin (RSPO) proteins belong to a family of secreted Wnt signalling agonists that binds LGR5 and form a complex with Frizzled and LRP to potentiate Wnt signalling97,98. The central player of the wnt pathway is beta-catenin, whose stability is controlled by the “destruction complex”. In absence of wnt receptor activation, Axin and the tumor suppressors adenomatous polyposis coli (APC) proteins form a scaffold that facilitates newly synthesized beta-catenin phosphorylation by two kinases, CK1α and GSK3β. The phosphorylated beta-catenin is subsequently ubiquitinylated, and undergoes proteasomal degradation. In the absence of wnt signalling, the levels of beta-catenin will decrease which allows the DNA binding protein Tcf/Lef to repress target gene expression by associating with co-repressors. However, when wnt receptors are engaged, the destruction complex is inactivated through relocation of Axin to the membrane. Beta-catenins can accumulate and enter into the nucleus, which transiently converts Tcf/Lef factors into transcription factors. LGR5 activation by R-spondins has been shown to enhance the WNT/beta-catenin signalling pathway leading to transcription of Tcf/Lef target genes98. Wnt/beta-catenin signalling also plays a key role in the development of glioma99. Persistent activation of beta-catenin has been found in the most malignant form of glioma GBM100. In addition, Wnt2 and Wnt5a are also overexpressed in human glioma. Knockdown of Wnt2 by siRNA in U251MG could inhibit cell proliferation101 whereas overexpression of Wnt5a 28 increases glioma cell proliferation in vitro102. However, the molecular mechanisms behind Wnt-LGR5 signalling in GBM development are not well understood. Figure 5. LGR5 and wnt signalling pathway. LGR5 activation by R-spondins has been shown to enhance the WNT/beta-catenin signalling pathway. Modified from103. LGR5 in cancer LGR5 expression is not only enriched in healthy human intestinal stem cells but also in tumor-initiation cells in human colorectal cancer. LGR5+ intestinal stem cells combined with loss of APC resulted in a progressively growing neoplasia104. LGR5 has further been validated as a bona-fide CSC marker identifying a fraction of colorectal cancer (CRC) cells with clonogenic and tumorigenic capacities. In stomach cancer, LGR5+ pyloric stem cells combined with loss of APC also lead to initiation of adenoma growth in the pylorus105. LGR5+ pyloric stem cells are suggested as the cell of origin of wnt-driven stomach cancer. In hair follicles, hundreds of regeneration cycles occur in a lifetime. In the resting phase of this cycle, LGR5+ cells are demonstrated to reside exclusive in secondary germ of telogen hair follicles 29 and the lower bulge that is/are believed to be the major stem cell reservoir for hair follicles106. Further, LGR5+ hair follicle stem cells are identified as a potential cell of origin for basal cell carcinoma107. Similar with intestine and stomach, these LGR5+ stem cells are potential targets for oncogenic mutations as a route to skin cancer. Recently, LGR5+ stem cells are shown to be radiosensitive and suggested to have the ability to overtime give rise to a radioresistant stem cell population in colorectal cancer108. LGR5 in GBM Regarding the role of LGR5 in gliomagenesis, this is poorly investigated with few reports so far. In 2013, Susumu et al. first demonstrated LGR5 as a marker of poor prognosis in GBM based on immunochemistry staining of a glioma tissue microarray (TMA) containing 283 different astrocytic tumors. LGR5 knockdown in one glioblastoma cell line leads to loss of sphere formation and increased apoptosis109. LGR5 was then revealed to be preferentially expressed in GBMs of the proneural subtype and could be regulated by the proneural factor OLIG2 through investigating two glioma cells lines with siRNA-mediated OLIG2 depletion110. LGR5 depletion in U87MG cells that cultured in serum containing medium, suspended sphere formation in vitro and tumor growth in vivo111.Recently, SOX9 is found to directly upregulate LGR5 mRNA expression by studying two glioblastoma cell lines112. And trichosanthin (a bioactive protein purified form tuberous root of a Chinese medical plant called Trichosanthes kirilowii) is discovered to induce apoptosis in U87 and U251 by targeting LGR5 and inhibiting the Wnt/beta-catenin signalling pathway113. 30 Present investigations Paper I. The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes Background Glioblastomas (GBMs) are highly heterogeneous and have recently have been classified into distinct molecular subtypes. For relevant in vitro and in vivo modeling of glioblastoma it is essential that the intra- and inter-tumor heterogeneity can be maintained. There is a general agreement that classical serum-derived cancer cell lines do not well represent the patients' tumors, and that defined serum-free stem cell culture conditions is an efficient method to enrich for glioma-initiating cells in culture. Aim To establish a large collection of relevant and well-characterized cell lines derived from GBM patients, maintained under serum-free stem cell culture conditions. Results and discussion In this study, 94 GBM surgical specimens were explanted in defined serumfree media and 53 GC lines were established. The dissociated tumors cells were first cultured as spheres and after 5-7 days transferred to laminincoated dish for adherent monolayer culture. Successful establishment of a GC line from a patient’s tumor is positively correlated with a short patient survival. To evaluate the proliferative capacity of these GC lines, we measured the cell density on day 1, day 4 and day 7. Patients whose tumors generated low proliferative cells exhibited better survival. However, a Cox multivariate analysis of several parameters (ability to propagated in culture, proliferative capacity and age) revealed that only age was a significant predictor of survival. Global gene expression followed by TCGA subtype classification was applied on 48 of the GC lines. In a subset of tumor samples, we analysed the molecular subtype of both primary tumors and their corresponding GC lines employing Nanostring technology. Due to intratumor heterogeneity and clonal selection during culturing, 10 of 22 cases the primary tissues and their 31 corresponding GC lines were assigned to the same molecular subtype. As GCIMP+ and IDH1 mutant proneural GBM patients showed better survival11, we also analysed if there was IDH1 mutations in our GC lines by exsome sequencing. We did not find the most frequent mutated site codon R132 in IDH1 in any of the analysed GC lines, and in support with a previous report showed that G-CIMP+ and IDH1 mutant GCs survival poorly in vitro114. The above findings indicated that all our GCs are G-CIMP-. When transplanting a majority of the GC lines to NOD-SCID mice, proneural GC lines displayed the shortest survival compared to the other subtypes. This goes well in line with a reduced survival of G-CIMP- proneural patients11. To investigate the genomic similarity to TCGA GBM cohort, analysis of copy number aberration was performed on 48 GC lines. Chromosome 7 gain and chromosome 10 loss were presented in more than 70% of the GC lines which was very similar to that of the tumors in TCGA cohort. To summarize, we have established a large collection of human GBM cells and these cells lines are tumorigenic, harbour genomic alterations characteristic of GBMs and represent all four TCGA molecular subtypes. This collection we refer to as the Human Glioblastoma Cell Culture (HGCC) resource, enable accurate cell-based modelling GBM and is connected with a database (www.hgcc.se) that provides high-resolution molecular data and clinical data. The HGCC resource provides an open-source repository for in vitro and in vivo modelling of GBM heterogeneity that will enable stratified studies of GBM mechanisms and facilitate the development of new therapies. Paper II. Malignancy and drug sensitivity of glioblastoma cells are affected by the cell of origin Background GBMs harbours highly intra-and inter-tumor heterogeneity and can be divided into molecular subtypes but these have to date no clinical relevance. The cell of origin for GBM is undefined but assumed to be a glial stem or progenitor cell. GBMs contain a subpopulation of cells called GSCs that has increased resistance to therapy and is believed to be responsible for tumor maintenance, progression and recurrence. Aim To investigate and cross-compare the consequences of the differentiation state of the cell of origin for GBM development and GBM cell properties in mouse and human cells. To identify a mouse cell origin (MCO) signature 32 and explore if that could be used to stratify GBM patients and find novel targets and pathways of importance for GBM development. Results and discussion In this paper, we used RCAS/tv-a mouse models of glioma to investigate how different states of differentiation of the initially transformed cell would affect GBM development. To induce glioma, three different tv-a expressing mouse strains (G/tv-a, N/tv-a and C/tv-a) were used to direct RCAS containing oncogene PDGF-B to infect GFAP, NES or CNP positive cells. All tv-a mouse lines were used in a homozygous Arf deficient background. To define the target cells in CNS, we initially injected DF-1 cells producing RCAS-eGFP in three tv-a lines at different locations in the brain. To target NSCs and OPCs, we injected adult G/tv-a;Arf-/- and C/tv-a;Arf-/mice in the SVZ. To increase the likelihood to target other types of glial precursor cells, we also inject N/tv-a;Arf-/- mice in the retrosplenial cortex (CTX). By co-immunofluorescence staining for neural/glial markers and GFP, we found RCAS infected CNS cells in G/tv-a SVZ brains displayed a more immature phenotype (GFAP+/NES+/CNP-/OLIG2-/SOX2+), a putatively NSC; in N/tv-a CTX brains displayed a glial precursor cell-like phenotype (GFAP+/NES+/CNP-/OLIG2+/SOX2+) and in C/tv-a SVZ brains the infected cells were similar to an OPC (GFAP-/NES-/CNP+/OLIG2+/ SOX2+). Regarding the mouse survival, G/tv-a SVZ and N/tv-a CTX tended to be more malignant than C/tv-a SVZ mice when DF-1 cells producing RCASPDGF-B were injected. Further, G/tv-a SVZ and N/tv-a CTX gave rise to a glioma incidence of 100%, while tumors generated in C/tv-a SVZ mice had an incidence of 82.6%. Subsequently, we cultured GBM cells (GCs) in serum-free defined NSC medium. The GC lines were named mGC1GFAP, mGC2NES and mGC3CNP for mouse GCs originating from G/tv-a SVZ, N/tv-a CTX and C/tv-a SVZ. We found mGC1GFAP cells of putative NSC origin were more tumorigenic and had higher self-renewal abilities than mGC2NES or mGC3CNP cells, i.e. of putative glial precursor cell origin. GCs of more immature origin are also remarkably more resistant to proliferative arrest in response to seruminduced differentiation and were more sensitive to drugs compared to a glial precursor cell origin. By transcriptome analysis we have generated a MCO gene signature that by cross-species bioinformatics could be used to stratify 64 human GCs and 529 GBM tissue samples. Human GCs in the mGC1GFAP group (=hGC1) showed higher self-renewal and tumorigenic capacities while being more sensitive to drugs compared to those assigned to the mGC2NES group (=hGC2). Here we show a functional relationship between the differentiation state of the initially transformed cell and the phenotype of GBM cells generated 33 thereof that could be applied on human GBMs, which could have important implications for therapy development and provide the basis for a new predictive MCO-based patient classification. Paper III. High LGR5 expression in proneural glioblastoma cells contributes to increased self-renewal and invasiveness Background Glioblastoma (GBM) maintenance, progression and recurrence are believed to be driven by GSCs that have unique/enhanced tumorigenic capacities and are more resistant to therapy. GSCs are characterized by increased selfrenewal capacity and have the ability to generate the full repertoire of tumor cells. LGR5 was discovered as a stem cell marker in several normal tissues and has been shown to both enhance and be induced by WNT signalling. Expression of LGR5 has also been found in various forms of cancer and has been validated as a cancer stem cell marker. In paper II, we identified LGR5 to be significantly upregulated in GBM cells (GCs) derived from tumors induced in GFAP-promoter active cells in the SVZ of adult mice, i.e. of putative NSC origin. Aim To analyse the role of LGR5 in GBM and its mechanism of action in GCs. Results and discussion By microarray analysis and subsequent qPCR validation LGR5 was found to be highly expressed in the most malignant mouse GC lines of putative NSC origin. Investigating our HGCC database we found that the expression of LGR5 was also significantly higher in the proneural subtype of human GBM, which is characterized by PDGFRA amplification/overexpression. When exploring LGR5 expression in TCGA database, LGR5 was also found to be enriched in the proneural subtype and a significant correlation between LGR5 and PDGFRA mRNA expression was discovered in both all GBMs (n=206) and in proneural GBM tumors (n=56). However, this positive correlation could not be detected when proneural samples were removed from the data set. To investigate the relation between LGR5 and PDGFRA, we made lentiviral vectors to be able to overexpress LGR5 in our GC lines. We found overexpression of LGR5 caused an upregulation of PDGFRA in LGR5high proneural GCs but not in LGR5low GCs by immunofluorescence staining. Co-expression of PDGFRA and LGR5 was observed in one of the LGR5high proneural GC lines. To study if PDGFRA expression would affect LGR5 expression, we treated LGR5high proneural GC lines with siPDGFRA, and 34 found that depletion of PDGFRA caused a significant decrease of LGR5 expression in two out of three GC lines. These findings suggest that LGR5 may play an important role in proneural GCs/GBM and there may be a functional connection between LGR5 and PDGFRA. By applying limiting dilution assay, we found LGR5high proneural GCs showed higher self-renewal capacity and generated larger spheres than LGR5high mesenchymal and LGR5low GCs. To investigate the functional consequence of high LGR5 expression in GCs, we made lentiviral constructs containing short hairpin-LGR5 (shLGR5) or a nonsense control shRNA (shNS). Depletion of LGR5 in LGR5high proneural GC lines had a hampering effect on self-renewal capacity. From our previous studies in mouse GCs we knew that self-renewal capacity is strongly correlated with tumorigenicity. By intracranially transplanting GC lines to NOD-SCID mice, we found LGR5high GC lines accelerated tumor development compared to LGR5low GC lines. As LGR5high proneural GCs had higher self-renewal ability than LGR5high mesenchymal GCs, we also transplanted these GCs into NOD-SCID mice and found that LGR5high proneural GCs were more tumorigenic than LGR5high mesenchymal GCs. To investigate if overexpression of LGR5 would affect the in vivo tumorigenic phenotype of GCs, we transplanted LGR5 overexpression GCs and corresponding controls into NOD-SCID mice. Overexpression of LGR5 in LGR5high proneural GCs caused both larger and invasive tumors compared to their controls. However, we did not observe this effect in LGR5low GC lines. Continually, we investigated the role of LGR5 in migration and invasion in vitro. We found depletion of LGR5 in LGR5high proneural cells caused decreased migration by applying scratch assay and decreased invasion capacity by applying 3-D invasion assay in collagen gels. In lines with in vivo studies, overexpression of LGR5 in LGR5low GCs did not increase their invasion capacity in vitro. Several studies in CSCs have indicated a role of LGR5 in WNT signalling and RSPOs have been shown to bind LGR5 to potentiate WNT signalling. We found knock-down of LGR5 affected beta-catenin nuclear localization upon WNT3a treatment. RSPO1 was found to potentiate WNT signalling in a subset of GCs. Differential mRNA expression analyses of a large set of glioma relevant genes in relation to LGR5 high and low expression generated many candidates that may be further investigated in order to identify the underlying mechanism of LGR5 signalling. Three of the genes, CCND2, OLIG2 and DKK1, were further investigated and all were found to be regulated by LGR5. Our findings point to an important role for LGR5 in sustaining stemness, invasiveness and tumorigenicity in proneural GCs, which makes it a putative predictive biomarker and a potential target for future drug development. 35 Paper IV. Oncogenic signalling is dominant to cell of origin and dictates astrocytic or oligodendroglial tumor development from oligodendrocyte precursor cells Background The majority of gliomas of grades II-IV is composed of astrocytic and oligodendroglial tumors, which are the two most common types of glioma that mainly affect adult. Astrocytic and oligodendroglial gliomas have been considered as separate disease groups with different diagnosis and putatively different origins. Oligodendrocyte precursor cells (OPCs) have been demonstrated as the cell of origin for experimental oligodendroglial tumors. While, if OPCs could be the cell of origin for experimental astrocytic gliomas remains largely unknown. Aim To investigate the potential and circumstance of OPCs to develop both astrocytic and oligodendroglial gliomas. Results and discussion In our lab, the Ctv-a mouse model was developed to study the role of OPCs in glioma development48. Thus it is the evidence that OPCs can be the cell origin for oligodendroglial-like tumor in mice. We wondered if OPCs also held the potential to develop astrocytomas. We then injected K-RAS+AKT into neonatal Ctv-a mice deficient for ink4a, Arf or ink4a-Arf. Both Ctv-a wild-type and ink4a-/- mice were not susceptible to K-RAS+AKT-induced glioma development. While Arf -/- and ink4a-Arf -/- mice could give rise to tumor development from OPCs with an incidence of 30% and 19% respectively. Quite interestingly, K-RAS+AKT-induced glioma from OPCs displayed an astrocytoma-like histopathology. To investigate if Cdkn2a loss was responsible for the astrocytic histopathology, we injected RCAS-PDGF-B into neonatal Ctv-a mice deficient for Arf or ink4a-Arf. Loss of Arf or ink4a-Arf also accelerated PDGF-B induced glioma development and increased tumor incidence. However, the histopathology of these tumors was analogous to human oligodendroglioma, which was in concordance with PDGF-B induced glioma in Ctv-a wild-type mice. These findings indicate that OPCs can give rise to both astrocytic and oligodendroglial tumors, depending on the activated oncogenes and loss of Cdkn2a. Subsequently, we compared expression of various glial proteins in KRAS+AKT and PDGF-B-induced tumors. Nestin and GFAP displayed homogenous expression in all K-RAS+AKT induced gliomas, while GFAP only detected in reactive astrocytes in PDGF-B induced tumors. Further, 36 several astrocytic markers like vimentin, CD44 and YKL40 were all displayed higher expression in K-RAS+AKT induced gliomas. OPC makers like PDGFRα and OLIG2 were shown to give a higher, more homogenous expression in PDGF-B induced tumors. By analyzing the transcriptome data from TCGA database, there was a clear overlap between human astrocytic and oligodendroglial tumors that supported our mouse data. Our findings indicate that oncogenic signalling is dominant to cell of origin (COO) in affecting the histologic classification of gliomas. This may have implications for future therapy development since we have data (in paper II) that shows that COO affects therapy response of GCs. 37 Future perspective Glioblastoma (GBM) is the most common and lethal form of primary brain tumor, which displays remarkable intra-and inter-tumor heterogeneity. These histologically inseparable adult GBMs can be classified into at least four different TCGA molecular subtype: proneural. neural, classical and mesenchymal 9. In paper I, we have described the establishment and characterization of a biobank of 48 cell lines derived from GBM patients using stem cell culture conditions and these GC lines represent all four subtypes. As different samples or even individual cells from the same GBM patients can give different molecular subtypes12,13, similarly only 45% of GC lines showed identical transcriptome subtype as the tumor of origin. Besides the sample heterogeneity, the existence of non-neoplastic cells, like inflammatory cells, reactive astrocytes and vascular cells can also contribute to the subtype shift. It would be very valuable to develop a new cell culture method to also maintain nonneoplastic cells. Additionally, most mesenchymal GC lines did not generate macroscopic tumors within 20 weeks, however diffusely spread human GCs could be detected in most of the mouse brain. It would be interesting to further investigate these mesenchymal lines, whether they represent a phenotypically distinct subgroups and if those seemingly quiescent but clearly alive human GCs can initiate GBM in mice by giving enough time. In paper II, we have analyzed the GBM development and essential GC properties by comparing tumors induced by three putative originating cell types. We have found that a more immature, NSC-like origin significantly accelerated GBM development, increased the number of GCs in the primary tumor and enhanced their malignant properties compared to a glial precursor cell origin. By transcriptome analysis we generated 196 mouse cell origin (MCO) gene signature to stratify human GBM cells and then performed drug test on the stratified groups. Interesting, both mouse and human GCs with higher self-renewal and tumorigenic capacity also displayed a greater drug sensitivity. To develop more effective drug treatment, we need to further investigate the mechanism of interplay between stemness and drug sensitivity. To further explore the MCO gene signature is also a way to reveal more candidate genes or pathways to contribute to GBM therapy. Additionally, when related our mouse GBM models to TCGA subtypes, it is clearly showed that the three mouse models could not cover the whole human GBM 38 diversity, which encourage both us and others to add more relevant mouse models and oncogenes to investigate cell of origin. In paper III, we investigated the role of LGR5 in sustaining stemness and invasive properties in proneural GCs. We found that LGR5 is highly enriched in the proneural subtype and correlated with proneural markers, like PDGFRA and OLIG2. It would be interesting to further explore the role of LGR5 in PDGFRA and p53 signalling. LGR5 did not affect proliferation but both self-renewal and invasive capacity of proneural GCs, which suggests it could serve as a potential therapeutic GBM target. By relating the expression of 758 glioma-relevant genes to high versus low LGR5 expression, we found LPAR4, CCND2, PDGFRA and OLIG2 were most up-regulated expressed in LGR5high GCs and DKK1 was significantly down-regulated in LGR5low GCs. Next, we will investigate more on these downstream genes of LGR5 and study their function connections with LGR5 and how they affect LGR5 signalling. In paper IV, we showed that human astrocytic and oligodendroglial tumors could have the same cell of origin and genetic alterations is dominant to cell of origin in determining glioma histopathology. Cell of origin can also affect the molecular subtype of GBMs, which is supported by us (Paper II) and others72. We also found that OPCs of neonatal mice were easier to generate PDGF-B induced glioma compared to OPCs in adult mice. This suggests that development age plays an essential role in glioma development. It would be interesting to further explore how a more immature origin influent glioma development in neonatal mice compared to adult mice. Gene expression profiles of neonatal and adult glioma will add values to further investigate the development age in gliomagenesis. In all, high-grade glioma is a very complex, heterogeneous and lethal disease that currently has no cure. It is imperative to stratify glioma patients into clinical relevant groups and identify effective drug targets for each subgroup of patients. By establishing a biobank of glioma patients derived GCs to model GBM and investigating cell of origin, genetic alterations and role of a cancer stem cell marker LGR5 in gliomagenesis, hopefully these investigations will contribute to the new therapy development and benefit glioma patients. 39 Acknowledgements This work has been carried out at the Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden. Financial support was provided by grants from the Swedish Cancer Society, the Swedish Research Council, the Swedish Childhood Cancer Foundation, Ragnar Söderberg's Foundation, Knut and Alice Wallenberg's Foundation, the Swedish Society of Medicine, the Faculty of Medicine and the Department of IGP at Uppsala University. I would like to take this opportunity to express my gratitude to the people who have supported and helped me during my work. My supervisor Lene Uhrbom, for accepting me as a PhD student in your research group, for guiding me in the scientific journey with patience, for supporting me in my life with your affection, for smiling and sharing happiness within our group, for always being there whenever I turned to you. My co-supervisor Bengt Westermark, for your profound knowledge and humorous way of expression. I am always impressed by your speaking either in conferences or in discussions, which encourages me to think more and deeper! My co-supervisor Anna Segerman, for motivating me with your enthusiasm in science, for sharing valuable ideas with me. All my co-authors and collaborators, especially, Yiwen, you are another cosupervisor for me. Your support throughout my life in Rudbeck at both scientific and personal level has been very helpful. E-Jean, you taught me how to make a fancy presentation and it is always joyful to talk and work with you. Usha, you are such a nice girl and thanks for all your helps and supports in the lab. Anders, for all the bioinformatics analysis you have done and I am very impressed by your quick response, efficient work and also your chocolate with 100% COCO. Tobias, for the nice collaboration on both HGCC and KAW projects. You are also a genius in fixing computer problem. Patrik, for establishing a searchable database for our HGCC resource and it is very glad to collaborate with you. Sven and Voichita, I am so lucky that you are here, my studies could not have moved into a further level without your invaluable advice and great contributions. Fredrik, there always a 40 smile on your face; always a brilliant idea from your mouth. Thanks for your invaluable advices and sharing the materials for molecular cloning. Karin, for all your encouragement and great supports. Chuan, for the knowledge and resources of lentivirus production. You are always there when I need you. Andries and Lei, for help me to detect endogenous protein level of LGR5 in GCs using in situ PLA. Marianne, you are the sunshine in our lab. You make the working environment so nice and warm. You are the best technician that I have ever seen. Smitha, you taught me how to do molecular cloning and how to lead a happy lifeJ. Sathish, for many helps in both cell lab and our benches. Malin T., for your hard working and you are one of the best students that I have ever supervised. Mia, for your sweet smile and many helps. Demet, for teaching me how to do stereotactic injections for newborn mice, and happiness we were sharing together. Nanna, still remember the time we spent together in New York. You are such a nice person. I will never forget you, a lovely pink girl. My past and present office mates: E-Jean, Vasil, Gabriela, Sara, Usha, Antonia, Lucy, Anna Sjösten, Demet, Anqi, Sanna, Vikki, Sathish, Ludmila, Smitha and Yiwen. Thank you for the innumerable laughs, biscuits, and sharing interesting experience. You make my life colorful. The past and present members of the rest of the neuro-oncology group: Elena, Jelena, Sanaz, Ananya, Annika, Soumi, Andreas, Lulu, Argyris, Grzegorz, Elin, Evgenia, Lukasz, Cecilia, Ingrid, Linnea, Anna Borgenvik, Matko, Sanna, Sonjia, Holger, Lisa, Hanna, Ida, Maria Ferletta, Erika, Malin Berglund, Malin Jarvius, Jacob, Ann Westermark, Anna Dimberg, Maria Georanaki, Luuk, Hua, Lei Zhang, Kalyani and Roberta for all the care and great moment in the lab. To the students that I have supervised: Malin Engvall, Lilian Kempe, Malin Tirfling, Augusta Broome and Tina Saren, for your contributions to the progress of LGR5 project. Special thank goes to Di, for many helps in molecular cloning, lentivirus production, computer problems and personal matters. I would not come to Sweden without your beautiful presentation of an introduction to Uppsala University. Lei Zhang, for the helps in qPT-PCR, IVIS machine and a very warm welcome when the first time I arrived in Uppsala. Pacholsky and Sara Petersson, for the helps in cell sorting. Matyas, for the helps in bioimaging. 41 Chirstina Magnusson, you always try your best to help me. The past and present guys and girls from the “Chinese lunch-table”: Xiujuan, Yiwen, Anqi, Dan, Hua, Lei Zhang, Hanqian, Liqun, Yan Zhang, Lei Chen, Kun Wang, Junhong, Di Wu, Jin Zhao and Yunyuan for the relaxing and delightful lunch time. To my buddies in Sweden, Kun Wei, Rui Miao, Henrik, Yinghua, Anders Lundqvist, Jing Ma, Lu Zhang, Xi Chen, Di Yu and Chuan Jin, for the difficulties, happiness and gossip we are sharing together. My life would have been boring without your accompany. To all my friends else where, especially to Lu Li, Hualian, Chunyu, Yiwen Liang and Guangting for caring about me and supporting me. Mum and Dad, for always loving, encouraging and believing in me all the time. 爸爸妈妈,谢谢你们爱我,鼓励我,相信我直到永远。 My dear Hao, thank you for always loving and supporting me, for always tolerating my bad tempers (seems only to youJ). Looking forward to the third member in our family! 42 References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. Ostrom, Q.T., et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neurooncology 16 Suppl 4, iv1-63 (2014). Furnari, F.B., et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes & development 21, 2683-2710 (2007). Louis, D.N., Ohgaki,H., Wiestler, O.D. & Cavenee, W.K. . WHO classification of tumors of the central nervous system. International Agency for research on Cancer (IARC) (2007). Reardon, D.A., Rich, J.N., Friedman, H.S. & Bigner, D.D. Recent advances in the treatment of malignant astrocytoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 24, 1253-1265 (2006). Johnson, D.R. & O'Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. Journal of neuro-oncology 107, 359364 (2012). Maher, E.A., et al. Malignant glioma: genetics and biology of a grave matter. Genes & development 15, 1311-1333 (2001). Sanai, N., Alvarez-Buylla, A. & Berger, M.S. Neural stem cells and the origin of gliomas. The New England journal of medicine 353, 811-822 (2005). Phillips, H.S., et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell 9, 157-173 (2006). Verhaak, R.G., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell 17, 98-110 (2010). Noushmehr, H., et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell 17, 510-522 (2010). Brennan, C.W., et al. The Somatic Genomic Landscape of Glioblastoma. Cell 155, 462-477 (2013). Sottoriva, A., et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. P Natl Acad Sci USA 110, 4009-4014 (2013). Patel, A.P., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396-1401 (2014). Meyer, M., et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. P Natl Acad Sci USA 112, 851-856 (2015). Sturm, D., et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer cell 22, 425-437 (2012). Gallo, M., et al. MLL5 Orchestrates a Cancer Self-Renewal State by Repressing the Histone Variant H3.3 and Globally Reorganizing Chromatin. Cancer cell 28, 715-729 (2015). 43 17. TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061-1068 (2008). 18. Heldin, C.H. & Westermark, B. Platelet-derived growth factor: mechanism of action and possible in vivo function. Cell regulation 1, 555-566 (1990). 19. Ohgaki, H. & Kleihues, P. Genetic pathways to primary and secondary glioblastoma. The American journal of pathology 170, 1445-1453 (2007). 20. Andrae, J., Gallini, R. & Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes & development 22, 1276-1312 (2008). 21. Noble, M., Murray, K., Stroobant, P., Waterfield, M.D. & Riddle, P. PlateletDerived Growth-Factor Promotes Division and Motility and Inhibits Premature Differentiation of the Oligodendrocyte Type-2 Astrocyte Progenitor-Cell. Nature 333, 560-562 (1988). 22. Raff, M.C., Lillien, L.E., Richardson, W.D., Burne, J.F. & Noble, M.D. Platelet-Derived Growth-Factor from Astrocytes Drives the Clock That Times Oligodendrocyte Development in Culture. Nature 333, 562-565 (1988). 23. Richardson, W.D., Pringle, N., Mosley, M.J., Westermark, B. & Duboisdalcq, M. A Role for Platelet-Derived Growth-Factor in Normal Gliogenesis in the Central Nervous-System. Cell 53, 309-319 (1988). 24. Calver, A.R., et al. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron 20, 869-882 (1998). 25. Nait Oumesmar, B., Vignais, L. & Baron-Van Evercooren, A. Developmental expression of platelet-derived growth factor alpha-receptor in neurons and glial cells of the mouse CNS. The Journal of neuroscience : the official journal of the Society for Neuroscience 17, 125-139 (1997). 26. Pringle, N.P., Mudhar, H.S., Collarini, E.J. & Richardson, W.D. PDGF receptors in the rat CNS: during late neurogenesis, PDGF alpha-receptor expression appears to be restricted to glial cells of the oligodendrocyte lineage. Development 115, 535-551 (1992). 27. Nister, M., Claesson-Welsh, L., Eriksson, A., Heldin, C.H. & Westermark, B. Differential expression of platelet-derived growth factor receptors in human malignant glioma cell lines. The Journal of biological chemistry 266, 1675516763 (1991). 28. Hermanson, M., et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer research 52, 3213-3219 (1992). 29. Guha, A., Dashner, K., Black, P.M., Wagner, J.A. & Stiles, C.D. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. International journal of cancer. Journal international du cancer 60, 168-173 (1995). 30. Lokker, N.A., Sullivan, C.M., Hollenbach, S.J., Israel, M.A. & Giese, N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer research 62, 3729-3735 (2002). 31. Schaudies, R.P., Christian, E.L. & Savage, C.R. Epidermal Growth-Factor Immunoreactive Material in the Rat-Brain - Localization and Identification of Multiple Species. Journal of Biological Chemistry 264, 10447-10450 (1989). 32. Yamada, M., Ikeuchi, T. & Hatanaka, H. The neurotrophic action and signalling of epidermal growth factor. Prog Neurobiol 51, 19-37 (1997). 33. Frederick, L., Wang, X.Y., Eley, G. & James, C.D. Diversity and frequency of Epidermal Growth Factor Receptor mutations in human glioblastomas. Cancer research 60, 1383-1387 (2000). 44 34. Gan, H.K., Kaye, A.H. & Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci 16, 748-754 (2009). 35. Dai, C.K. & Holland, E.C. Glioma models. Bba-Rev Cancer 1551, M19-M27 (2001). 36. Tator, C.H., Day, A., Ng, R. & Liberman, L. Chemotherapy of an Experimental Glioma with Nitrosoureas. Cancer research 37, 476-481 (1977). 37. Rushing, E.J., Watson, M.L., Schold, S.C., Land, K.J. & Kokkinakis, D.M. Glial tumors in the MNU rat model: Induction of pure and mixed gliomas that do not require typical missense mutations of p53. J Neuropath Exp Neur 57, 1053-1060 (1998). 38. Barth, R.F. Rat brain tumor models in experimental neuro-oncology: The 9L, C6, T9, F98, RG2 (D74), RT-2 and CNS-1 gliomas. J Neuro-Oncol 36, 91-102 (1998). 39. Plate, K.H., Breier, G., Millauer, B., Ullrich, A. & Risau, W. Up-Regulation of Vascular Endothelial Growth-Factor and Its Cognate Receptors in a Rat Glioma Model of Tumor Angiogenesis. Cancer research 53, 5822-5827 (1993). 40. Kondo, S., Kondo, Y., Li, G., Silverman, R.H. & Cowell, J.K. Targeted therapy of human malignant glioma in a mouse model by 2-5A antisense directed against telomerase RNA. Oncogene 16, 3323-3330 (1998). 41. Holland, E.C. Gliomagenesis: genetic alterations and mouse models. Nature reviews. Genetics 2, 120-129 (2001). 42. Dale, E.C. & Ow, D.W. Gene-Transfer with Subsequent Removal of the Selection Gene from the Host Genome. P Natl Acad Sci USA 88, 10558-10562 (1991). 43. Uhrbom, L. & Holland, E.C. Modeling gliomagenesis with somatic cell gene transfer using retroviral vectors. J Neuro-Oncol 53, 297-305 (2001). 44. Khaled, W.T. & Liu, P. Cancer mouse models: past, present and future. Semin Cell Dev Biol 27, 54-60 (2014). 45. Zuckermann, M., et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun 6, 7391 (2015). 46. Holland, E.C. & Varmus, H.E. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. P Natl Acad Sci USA 95, 1218-1223 (1998). 47. Holland, E.C., Hively, W.P., DePinho, R.A. & Varmus, H.E. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cellcycle arrest pathways to induce glioma-like lesions in mice. Genes & development 12, 3675-3685 (1998). 48. Lindberg, N., Kastemar, M., Olofsson, T., Smits, A. & Uhrbom, L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 28, 2266-2275 (2009). 49. Schuller, U., et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer cell 14, 123-134 (2008). 50. Vervoort, V.S., et al. A novel Flk1-TVA transgenic mouse model for gene delivery to angiogenic vasculature. Transgenic research 17, 403-415 (2008). 51. Morton, J.P., et al. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. P Natl Acad Sci USA 104, 5103-5108 (2007). 52. Montaner, S., et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer cell 3, 23-36 (2003). 45 53. Dai, C., et al. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & development 15, 1913-1925 (2001). 54. Dunlap, S.M., et al. Insulin-like growth factor binding protein 2 promotes glioma development and progression. P Natl Acad Sci USA 104, 11736-11741 (2007). 55. Tchougounova, E., et al. Loss of Arf causes tumor progression of PDGFBinduced oligodendroglioma. Oncogene 26, 6289-6296 (2007). 56. Uhrbom, L., Kastemar, M., Johansson, F.K., Westermark, B. & Holland, E.C. Cell type-specific tumor suppression by Ink4a and Arf in kras-induced mouse gliomagenesis. Cancer research 65, 2065-2069 (2005). 57. Doetsch, F., Caillé, I., Lim, D.A., García-Verdugo, J.M. & Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703-716 (1999). 58. Barami, K., et al. Relationship of gliomas to the ventricular walls. J Clin Neurosci 16, 195-201 (2009). 59. Zappone, M.V., et al. Sox2 regulatory sequences direct expression of a (beta)geo transgene to telencephalic neural stem cells and precursors of the mouse embryo, revealing regionalization of gene expression in CNS stem cells. Development (Cambridge, England) 127, 2367-2382 (2000). 60. Reynolds, B.A. & Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (New York, N.Y.) 255, 1707-1710 (1992). 61. Uchida, N., et al. Direct isolation of human central nervous system stem cells. Proceedings of the National Academy of Sciences of the United States of America 97, 14720-14725 (2000). 62. Roelofs, R.F., et al. Adult human subventricular, subgranular, and subpial zones contain astrocytes with a specialized intermediate filament cytoskeleton. Glia 52, 289-300 (2005). 63. Zhu, Y., et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer cell 8, 119-130 (2005). 64. Alcantara Llaguno, S., et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer cell 15, 45-56 (2009). 65. Chen, J., et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522-526 (2012). 66. Alves, J.A.J., Barone, P., Engelender, S., Froes, M.M. & Menezes, J.R.L. Initial stages of radial glia astrocytic transformation in the early postnatal anterior subventricular zone. J Neurobiol 52, 251-265 (2002). 67. Lim, D.A. & Alvarez-Buylla, A. Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. P Natl Acad Sci USA 96, 7526-7531 (1999). 68. Xiao, A., Wu, H., Pandolfi, P.P., Louis, D.N. & Van Dyke, T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer cell 1, 157-168 (2002). 69. Friedmann-Morvinski, D., et al. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science 338, 1080-1084 (2012). 70. Persson, A.I., et al. Non-stem cell origin for oligodendroglioma. Cancer cell 18, 669-682 (2010). 46 71. Liu, C., et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209-221 (2011). 72. Alcantara Llaguno, S.R., et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer cell 28, 429-440 (2015). 73. Nowell, P.C. The clonal evolution of tumor cell populations. Science 194, 2328 (1976). 74. Baylin, S.B. & Jones, P.A. A decade of exploring the cancer epigenome biological and translational implications. Nature reviews. Cancer 11, 726-734 (2011). 75. Charles, N., et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell stem cell 6, 141-152 (2010). 76. Magee, J.A., Piskounova, E. & Morrison, S.J. Cancer Stem Cells: Impact, Heterogeneity, and Uncertainty. Cancer cell 21, 283-296 (2012). 77. Al-Hajj, M., Wicha, M.S., Benito-Hernandez, A., Morrison, S.J. & Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. P Natl Acad Sci USA 100, 3983-3988 (2003). 78. Lapidot, T., et al. A Cell Initiating Human Acute Myeloid-Leukemia after Transplantation into Scid Mice. Nature 367, 645-648 (1994). 79. Singh, S.K., et al. Identification of human brain tumour initiating cells. Nature 432, 396-401 (2004). 80. Visvader, J.E. & Lindeman, G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nature reviews. Cancer 8, 755-768 (2008). 81. Son, M.J., Woolard, K., Nam, D.H., Lee, J. & Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell stem cell 4, 440-452 (2009). 82. Singh, S.K., et al. Identification of a cancer stem cell in human brain tumors. Cancer research 63, 5821-5828 (2003). 83. Bao, S.D., et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756-760 (2006). 84. Wang, J., et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. International Journal of Cancer 122, 761-768 (2008). 85. Anido, J., et al. TGF beta receptor inhibitors target the CD44high/Id1high glioma stem cell population in human glioblastoma. Ejc Suppl 8, 10-11 (2010). 86. Bao, S.D., et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer research 68, 6043-6048 (2008). 87. Tchoghandjian, A., et al. A2B5 Cells from Human Glioblastoma have Cancer Stem Cell Properties. Brain Pathol 20, 211-221 (2010). 88. Ogden, A.T., et al. Identification of A2B5(+)CD133-tumor-initiating cells in adult human gliomas. Neurosurgery 62, 505-514 (2008). 89. Lathia, J.D., et al. Integrin Alpha 6 Regulates Glioblastoma Stem Cells. Cell stem cell 6, 421-432 (2010). 90. Ge, Y.Q., et al. Sox2 is translationally activated by eukaryotic initiation factor 4E in human glioma-initiating cells. Biochem Bioph Res Co 397, 711-717 (2010). 91. Guo, Y., et al. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology 59, 763-775 (2011). 92. Hagerstrand, D., et al. Identification of a SOX2-dependent subset of tumorand sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro-oncology 13, 1178-1191 (2011). 47 93. Bexell, D., Gunnarsson, S., Siesjo, P., Bengzon, J. & Darabi, A. CD133+and nestin plus tumor-initiating cells dominate in N29 and N32 experimental gliomas. International Journal of Cancer 125, 15-22 (2009). 94. Zhang, M.Y., et al. Nestin and CD133: valuable stem cell-specific markers for determining clinical outcome of glioma patients. J Exp Clin Canc Res 27(2008). 95. Du, Z.H., et al. Oct4 is Expressed in Human Gliomas and Promotes Colony Formation in Glioma Cells. Glia 57, 724-733 (2009). 96. Ikushima, H., et al. Glioma-initiating Cells Retain Their Tumorigenicity through Integration of the Sox Axis and Oct4 Protein. Journal of Biological Chemistry 286, 41434-41441 (2011). 97. de Lau, W., et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293-U257 (2011). 98. Carmon, K.S., Gong, X., Lin, Q.S., Thomas, A. & Liu, Q.Y. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proceedings of the National Academy of Sciences of the United States of America 108, 11452-11457 (2011). 99. Palos, T.P., Zheng, S. & Howard, B.D. Wnt signaling induces GLT-1 expression in rat C6 glioma cells. Journal of neurochemistry 73, 1012-1023 (1999). 100. Zheng, H., et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer cell 17, 497-509 (2010). 101. Pu, P., et al. Downregulation of Wnt2 and beta-catenin by siRNA suppresses malignant glioma cell growth. Cancer gene therapy 16, 351-361 (2009). 102. Yu, J.M., et al. Role of Wnt5a in the proliferation of human glioblastoma cells. Cancer letters 257, 172-181 (2007). 103. Eisenmann, D.M. Wnt signaling. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.7.1. (2005). 104. Barker, N., et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608-611 (2009). 105. Barker, N., et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6, 25-36 (2010). 106. Jaks, V., et al. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat Genet 40, 1291-1299 (2008). 107. Kasper, M., et al. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proceedings of the National Academy of Sciences of the United States of America 108, 4099-4104 (2011). 108. Asfaha, S., et al. Krt19(+)/Lgr5(-) Cells Are Radioresistant Cancer-Initiating Stem Cells in the Colon and Intestine. Cell Stem Cell 16, 627-638 (2015). 109. Nakata, S., et al. LGR5 is a Marker of Poor Prognosis in Glioblastoma and is Required for Survival of Brain Cancer Stem-Like Cells. Brain Pathol 23, 6072 (2013). 110. Mao, X.G., et al. LGR5 is a proneural factor and is regulated by OLIG2 in glioma stem-like cells. Cellular and molecular neurobiology 33, 851-865 (2013). 111. Wang, D., et al. Knockdown of LGR5 suppresses the proliferation of glioma cells in vitro and in vivo. Oncol Rep 31, 41-49 (2014). 112. Hiraoka, K., et al. SOX9-mediated upregulation of LGR5 is important for glioblastoma tumorigenicity. Biochem Bioph Res Co 460, 216-221 (2015). 113. Miao, J., et al. Trichosanthin suppresses the proliferation of glioma cells by inhibiting LGR5 expression and the Wnt/beta-catenin signaling pathway. Oncology reports 34, 2845-2852 (2015). 48 114. Balvers, R.K., et al. Serum-free culture success of glial tumors is related to specific molecular profiles and expression of extracellular matrix-associated gene modules. Neuro-oncology 15, 1684-1695 (2013). 49 Acta Universitatis Upsaliensis Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine Editor: The Dean of the Faculty of Medicine A doctoral dissertation from the Faculty of Medicine, Uppsala University, is usually a summary of a number of papers. A few copies of the complete dissertation are kept at major Swedish research libraries, while the summary alone is distributed internationally through the series Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine. (Prior to January, 2005, the series was published under the title “Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine”.) Distribution: publications.uu.se urn:nbn:se:uu:diva-278529 ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2016
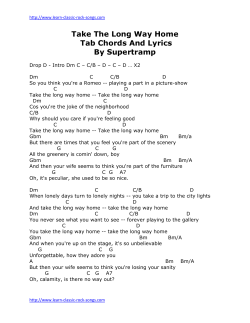
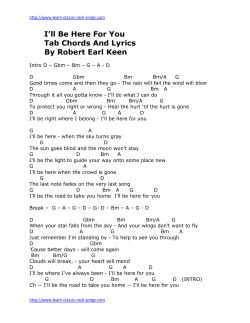








© Copyright 2025