
Tuesday, February 10
Tuesday, February 10, 2015 Symposium: Biophysics of RNA Processing: Degradation, Splicing, DEAD Box Proteins 1684-Symp Insights into Helicase Evolution from the Specificity and Mechanism of a Dead-Box Protein Anna L. Mallam, David J. Sidote, Alan M. Lambowitz. Molecular Biosciences and Chemsitry, University of Texas at Austin, Austin, TX, USA. How helicase families with a conserved catalytic ‘helicase core’ evolved to function on varied RNA and DNA substrates by diverse mechanisms remains unclear. Here, we used the helicase core of Mss116, a DEAD-box protein that utilizes ATP to locally unwind dsRNA, to investigate helicase specificity and mechanism. Previously, we found that the two RecA-like domains of the helicase core of Mss116 are in an extended ‘open state’ in the absence of substrates and recognize ATP and duplex RNA in a modular manner. Upon formation of a compact ‘closed state’ containing an ATPase active site, conserved motifs in the first domain promote the nonprocessive unwinding of short duplex substrates bound to the second domain by excluding one RNA strand and bending the other. In the present work, we define the molecular basis for the specificity of DEAD-box proteins. However, we also find that Mss116 has ambiguous substrate unwinding properties and interacts with a variety of NTPs and nucleic acids. The efficiency of unwinding correlates with the stability of the closed-state helicase core, a complex with nucleotide and nucleic acid that forms as duplexes are unwound. Crystal structures reveal that core stability is modulated by family-specific interactions that favor certain substrates. This suggests how present-day helicases diversified from an ancestral core with broad specificity by retaining core closure as a common catalytic mechanism while optimizing substrate-binding interactions for different cellular functions. 1685-Symp Single-Molecule Imaging of Pre-mRNA Splicing Sanjay Tyagi. Public Health Research Institute, Rutgers University, Newark, NJ, USA. We have developed a single-molecule RNA imaging method to study the intracellular sites of splicing. In this approach introns and exons in natural genes are probed in situ with distinctively labeled sets of about 50 oligonucleotide probes. The attachment of so many probes renders each target molecule so intensely fluorescent that it becomes visible as a bright diffraction-limited spot. Spots that fluoresce in just one color are either free introns or spliced mRNAs, and those that are visible in both colors correspond to pre-mRNA molecules. Our imaging studies confirm that constitutively spliced introns are removed at the gene locus during transcription. However, during alternative splicing events regulated by RNA binding proteins Sex lethal (in fruit flies) and polypyrimidine tract binding protein (in HeLa cells), which ensure that only one splice form is produced in a particular cell type, splicing gets uncoupled from transcription and occurs after the release of pre-mRNA from the gene locus. Similar uncoupling occurrs when intronic polypyrimidine tracts are masked within hairpins. Live cell imaging of the post-transcriptional splicing events suggests that they occur at the periphery of nuclear speckles where many splicing factors and poly adenylated transcripts are sequestered. Recent genome-wide studies reveal that a fraction of alternative splicing events likely occur via this route. 1686-Symp Auxiliary Factors and RNA Substrates Regulate Dead-Box Protein Activity by Modulation of the Dead-Box Protein Conformational Cycle Ulf Harms, Alexandra Z. Andreou, Airat Gubaev, Dagmar Klostermeier. Physical Chemistry, University of Munster, Munster, Germany. DEAD-box proteins unwind RNA duplexes at the expense of ATP hydrolysis in virtually all processes involving RNA. During unwinding, they alternate between open and closed conformations, and the transition to the closed conformation has previously been linked to duplex destabilization. The eukaryotic translation initiation factor 4A (eIF4A) is a DEAD-box helicase that is thought to resolve secondary structure elements from the 50 -UTR of mRNAs to enable ribosome scanning. Its RNA-stimulated ATPase and ATP-dependent helicase activities are enhanced by other translation initiation factors, but the underlying mechanisms are unclear. eIF4A can adopt three different conformations, an open state in the absence of ligands, a half-open state stabilized by eIF4G, and a closed state populated in the presence of eIF4G and eIF4B. In single molecule FRET experiments on donor/acceptor-labeled eIF4A, we have dissected the effect of eIF4B and eIF4G on RNA-dependent ATPase- and RNA helicase activities, and on the eIF4A conformational cycle in the context of different unwinding substrates. We show that eIF4B and eIF4G, as well as 335a different structures in the 50 -UTR, modulate the energy landscape underlying the eIF4A conformational cycle by changing the energetic differences and the energy barriers between functionally relevant conformational states, leading to changes in rate constants for inter-conversion and in equilibrium distributions. Our results reveal on a molecular level how translation initiation factors synergistically stimulate the eIF4A helicase activity in the mRNA scanning process, and show that the eIF4A conformational cycle is central for the multi-layered regulation of eIF4A activity, and for its role as a regulatory hub in translation initiation. 1687-Symp Molecular Mechanisms of Viral RNA Detection: RIG-I and MDA5 Sun Hur. Biological Chemistry and Molecular Pharmacology, Harvard University, Boston, MA, USA. The ability to distinguish ‘‘self’’ from ‘‘non-self’’ is fundamental to proper functioning of both the innate and adaptive immune systems. Several pattern recognition receptors (PRR) in the innate immune system are responsible for the initial detection of foreign molecules associated with pathogens such as bacterial cell wall components or viral nucleic acids. My laboratory has been interested in understanding the molecular mechanisms of one such family of receptors, RIG-I and MDA5, which recognize viral RNAs during infection and elicit the type I interferon response against a broad range of viruses. These receptors share the common domain architecture, including the DExD/H motif helicase domain. In this talk, I will present our recent discoveries on the structural and biochemical mechanisms by which these receptors differentially utilize the helicase domain to recognize their cognate viral RNAs and activate the signaling pathway. Symposium: Molecules of Memory: Glutamate Receptor Channels 1688-Symp Conformational Changes Underlying Glutamate Receptor Gating Mark Mayer. LCMN, NIH, Bethesda, MD, USA. The gating mechanism of glutamate receptor ion channels involves conformational changes triggered by the binding of glutamate that open the ion channel, followed within milliseconds by nearly complete desensitization. In a collaboration with the Subramaniam Lab at NCI, cryo-EM structures of full length AMPA (GluA2) receptors, stabilized in the active state by allosteric modulators, reveal concerted conformational changes in the ligand binding domain that produce a vertical contraction accompanied by a corkscrew like rotation that leads to expansion of the linkers leading to the M3 helix bundle; in the desensitized state the ATD dimer pairs adopts multiple conformations with evidence for substantial reorganization of the ligand binding domain tetramer. By contrast, for the GluK2 kainate receptor the ATD dimer assembly remains intact in the desensitized state, while the LBD rearranges into a 4-fold symmetric structure in which the linkers leading to the M3 helix bundle adopt a contracted conformation that is however different from the closed state. Measurements of ATD tetramer association by analytical ultracentrifugation reveal a 1000-fold high higher affinity for GluK2 versus GluA2 that underlies the different behavior of the ATD in the desensitized state, movements of which however are secondary to and play no major role in desensitization. 1689-Symp AMPA Receptor Structure, Function, and Dynamics Robert E. Oswald, Ahmed H. Ahmed, Christopher P. Ptak, Madeline Martinez. Molecular Medicine, Cornell University, Ithaca, NY, USA. AMPA receptors (GluAs) are essential neuronal ligand-gated ion channels involved in learning and memory. The dimeric conformation of the GluA ligand-binding domain is involved in the coupling of agonist binding to channel gating. We have used NMR, crystallography, ITC, and single channel recording to study the mechanism of action of antagonists and partial agonists on the GluA2 receptor. Antagonists form stable but open cleft binding sites with little dynamics, with binding driven largely by enthalpy. On the other hand, considerable dynamics are observed in the binding site in the presence of partial agonists, whose binding, in most cases, has a large entropic component. Allosteric modulators bind to a large surface that is formed by the dimer interface of two ligand-binding domains in the resting and channel activated states. This binding prevents the dissociation of the dimer interface and inhibits desensitization of the receptor. The desensitized conformation is disrupted along the dimer interface; however, little is known about the dynamic equilibrium between the 336a Tuesday, February 10, 2015 bound/dimerized form and the unbound/monomer forms. Using small angle x-ray scattering (SAXS), crystallography, and NMR spectroscopy, we developed an equilibrium model for modulator dependent dimerization. This model demonstrates that a second modulator-binding site produces both an increase in positive cooperativity and a higher apparent affinity. A combination of the crystal structures of the bound modulators and the binding model developed using SAXS data provide new clues for the development of more effective allosteric modulators that may have cognitive enhancing effects. 1690-Symp Intracellular Domains of NMDA Receptors Control Channel Permeation and Gating Properties Gabriela K. Popescu. Biochemistry, University at Buffalo (SUNY), Buffalo, NY, USA. Among glutamate-gated excitatory channels, NMDA receptors are pivotal to the physiology of central synapses. Their activation initiates cellular processes responsible for synaptic plasticity, the substrate of memory; but it can also awake apoptotic cascades that result in neuronal degeneration. Which cellular pathways are activated hinges critically on the amplitude and time course of the intracellular calcium injected by NMDA receptors. In turn, these features of the calcium transient depend fundamentally on the receptor’s ionic conductance, calcium permeability, and gating kinetics. Here, I present new evidence from my laboratory that the NMDA receptor-mediated calcium transient is differentially controlled by the receptor’s intracellular domains, with ionic conductance and calcium permeability set by GluN1 and gating kinetics controlled largely by GluN2 subunits. Importantly, the phosphorylation state of GluN1 residues modulates in a reversible and dynamic manner calcium permeability and pore size. This appears to be a heretofore unique case of physiologic regulation of channel permeability by reversible, in-situ post-translational modification. Given the critical role of NMDA receptor calcium transients in synaptic physiology, the mechanisms we discuss here may open the way for new ways to manipulate these for therapeutic gain. 1691-Symp NMDA Receptors as Dynamic Allosteric Machines Pierre Paoletti. Ecole Normale Superieure, CNRS, INSERM, Paris, France. N-methyl-D-aspartate receptors (NMDARs) are glutamate-gated ion channels that are essential mediators of excitatory neurotransmission and synaptic plasticity. NMDARs have also been implicated in a plethora of neuropathological conditions thus receiving strong interest as potential therapeutic targets. Recent years have witnessed major progress in our understanding of the structure, mechanisms and pharmacology of NMDARs, with highlights including the decoding of the first full-length receptor crystal structures and the discovery of complex allosteric interactions between the constitutive domains and subunits. NMDARs form massive (>600 kDa) heterotetrameric complexes that usually incorporate two obligatory GluN1 subunits and two GluN2 subunits, of which there are four subtypes (GluN2A-D). Here, I will present the unique role and structural mechanisms of the large extracellular N-terminal domains (NTDs). These domains are distinct from the agonist-binding domains and lay most distal from the transmembrane pore but have been shown to control receptor activity. I will highlight recent experimental and modeling data showing that the allosteric capacity of NMDAR NTDs is intimately linked to their high conformational mobility, the clamshell-like NTDs undergoing large scale motions that can be sensed by the downstream gating machinery. The NTD allosteric signaling in NMDARs is unique among the ionotropic glutamate receptor family with important implications both for receptor physiology and drug action. Platform: Single-Molecule Spectroscopy 1692-Plat NMDA Receptor Ion Channel Dynamics in Living Cells by a Novel Single-Molecule Patch-Clamp FRET Microscopy: Revealing the Multiple Conformational States Associated with a Channel at its Electrical Off State Dibyendu Sasmal, H. Peter Lu. Department of Chemistry and the Center for Photochemical Sciences, Bowling Green State University, Bowling Green, OH, USA. Stochastic and inhomogeneous conformational changes regulate the function and dynamics of ion channels. The conformational dynamics is often inhomogeneous and extremely difficult to be directly characterized by ensembleaveraged spectroscopic imaging or only by single channel patch-clamp electric recording methods. We have developed a new combined approaches of using single ion channel patch-clamp electrical recording and single-molecule fluorescence imaging for probing ion channel conformational changes simulta- neously with the electrical single channel recording. We were able to probe single ion-channel-protein conformational changes simultaneously with the electric on-off signals, and thus providing an understanding the dynamics and mechanism of ion-channel proteins at the molecular level.(1,2) We have probed NMDA (N-Methyl-D-Aspartate) receptor ion channel in live HEK293 cell, especially, the single ion channel open-close activity and its associated protein conformational changes simultaneously. Furthermore, we have revealed that the seemingly identical electrically off states are associated with multiple conformational states. Based on our experimental results, we have proposed a multistate clamshell model to interpret the NMDA receptor open-close dynamics. Our results shed light on new perspectives of the intrinsic interplay of lipid membrane dynamics, solvation dynamics, and the ion channel functions. Reference: 1. Dibyendu Kumar Sasmal, H. Peter Lu, ‘‘ Single-Molecule Patch-Clamp FRET Microscopy Studies of NMDA Receptor Ion Channel Dynamics in Living Cells: Revealing the Multiple Conformational States Associated with a Channel at Its Electrical Off State,’’ J. Am. Chem. Soc., 136, 12998-13005 (2014). 2. Suneth P. Rajapaksha, Xuefei Wang, H. Peter Lu, ‘‘Suspended Lipid Bilayer for Optical and Electrical measurements of Single Ion Channel Proteins,’’ Anal. Chem., 85, 8951-8955 (2013). 1693-Plat Exploring Tau Conformations at the Single-Molecule Level in a Microfluidic Trap Randall H. Goldsmith1, Sharla Wood1, Lydia Manger1, Michael Holden2, Martin Margittai2. 1 Chemistry, University of Wisconsin Madison, Madison, WI, USA, 2 Chemistry, University of Denver, Denver, CO, USA. The conformational dynamics of intrinsically disordered proteins (IDP’s) are inextricably linked to their roles in signaling, regulation, folding, and diseases. Single-molecule methods can contribute valuable information on the conformational dynamics of biomolecules because they allow the observation of unsynchronized dynamics and characterization of diverse populations. Typically, target biomolecules are immobilized to allow study over a longer time window. However, biomolecules with more fluid structures, like IDP’s, are highly susceptible to having their structure dominated by the immobilization environment. A method of studying single solution-phase biomolecules for prolonged periods of time would be highly useful for elucidating protein dynamics over many timescales. In this study, we present the use of a microfluidic trap that is capable of canceling Brownian motion to allow the observation of solution-phase dynamics of IDP’s over multiple seconds. We will focus on Tau, a protein contributor to the etiology of Alzheimer’s disease. Solution-phase conformations of the monomer and small aggregates will be described. The details of the technique, dynamics of the biomolecule targets, and future applications and directions will be discussed. 1694-Plat 3D Tracking of Single Quantum Dots through Off-Focus Imaging Lucia Gardini1,2, Marco Capitanio1,2, Francesco Saverio Pavone1,2. 1 LENS-University of Florence, Sesto Fiorentino, Italy, 2Physics DepartmentUniversity of Florence, Sesto Fiorentino, Italy. Recently, tremendous improvements have been achieved in the precision of localization of single fluorescent molecules, allowing localization and tracking of biomolecules at the nm level. Since the behaviour of proteins and biological molecules is tightly related to the cell’s environment, a growing number of microscopy techniques are moving from in vitro to live cell experiments. Looking at both diffusion and active transportation processes inside a cell requires threedimensional localization over a few microns range, high SNR images and high temporal resolution (ms order of magnitude). It has been shown that axial localization within few nanometers can be achieved through out-of-focus imaging, by studying the behaviour of the point spread function of probes out of the focal plane. Here we describe a new method, based on this approach, through which the x, y coordinates of the PSF’s centre are localized and the radius of the off-focus PSF is automatically measured and related to the axial position of the probe, thus providing a calibration curve for indirect axial position measurement. Our method revealed a non-linear behaviour of this curve for both fluorescent beads and quantum dots. Through our algorithm, simultaneous localization of all three dimensions within 5 nm accuracy can be achieved, over a 2 um range, for 200 nm fluorescent beads. Moreover, by the combination of off-focus imaging with HILO (Highly Inclined and Laminated Optical sheet) illumination we demonstrate 3D tracking of single QDs inside living cells within 10 nm accuracy over 1um range. Tuesday, February 10, 2015 1695-Plat Deconstructing PIFE: A Spectroscopic Investigation of the ‘‘Protein Induced Fluorescence Enhancement’’ Phenomenon in Cy3 Elana Maria Shepherd Stennett, Monika Anna Ciuba, Marcia Levitus. Arizona State University, Tempe, AZ, USA. PIFE, an acronym that stands for ‘‘Protein Induced Fluorescence Enhancement’’, is a term that has been coined to describe the enhancement of fluorescence intensity that the dye Cy3 experiences in the proximity of a protein. The approach has been used to study dynamic aspects of a large number of DNAand RNA-protein interactions at the single molecule level. We and others have hypothesized that the phenomenon results from a restriction in the photoisomerization deactivation pathway of the dye. Here, we present the results of a detailed spectroscopic study that aims to characterize PIFE at the molecular level. We used time-resolved fluorescence, fluorescence anisotropy and transient spectroscopy to fully characterize the deactivation pathways of the dye on DNA when next to a protein. Our results allowed us to confirm our hypothesis that the enhancement in fluorescence correlates with a decreased yield of photoisomer. 1696-Plat Fabrication and Surface Functionalization of Highly Birefringent Rutile Particles for Trapping in an Optical Torque Wrench Seungkyu Ha, Yera Ussembayev, Richard Janissen, Maarten van Oene, Nynke H. Dekker. Bionanoscience, TU Delft, Delft, Netherlands. The optical torque wrench (OTW) allows the direct application and measurement of torque on biomolecules, such as DNA or DNA-protein complexes, or rotary motors like the F0F1-ATP-synthase or the bacterial flagellar motor. The applicable torque of the OTW is a function of the size and birefringence of the particle. Quartz has proven a convenient material, but its quite low birefringence limits full investigation of torque-speed relationships of diverse biological systems. In contrast, rutile exhibits a much higher birefringence exceeding that of quartz by a factor of 30 - but its utilization has been infrequent because of the difficulties in optical trapping and fabrication. To enhance the applicability of the OTW, we have improved both the design and fabrication of cylindrical rutile particles. We have employed finite element method calculations to determine the optimal dimension of stably trappable rutile cylinders. To obtain rutile cylinders with the optimal dimensions, we developed a protocol for full control of size and sidewall angle. In our fabrication protocol, a chromium etch mask provides increased resistance to dry etching and allows the fabrication of structures with both high aspect ratio and anisotropy. Also, the sidewall angle of cylinders can be readily tuned by adjusting a single process parameter, namely the oxygen flow rate during dry etching. The fabricated cylinders were characterized in the OTW setup to reveal their linear and angular trapping properties. The fabrication process is compatible with common chemical functionalization procedures and permits covalent biomolecule attachment. To enhance biomolecule coverage, we used ethanolamine and poly(ethylene glycol) as biomolecular crosslinkers to obtain homogenous and dense coatings. Our recent results, in which we use functionalized, trapped rutile cylinders to study single biomolecules and motor proteins, will be presented. 1697-Plat Electron Paramagnetic Resonance from a Single Biomolecule Richelle M. Teeling-Smith1, Young Woo Jung2, Nicolas J. Scozzaro1, Jeremy Cardellino1, Isaac Rampersaud3, Justin A. North1, Marek Simon1, Vidya P. Bhallamudi1, Arfaan Rampersaud3, Ezekiel Johnston-Halperin1, Michael G. Poirier1, P. Chris Hammel1. 1 Physics, The Ohio State University, Columbus, OH, USA, 2Samsung Electronics, Yongin-City, Korea, Republic of, 3Columbus Nanoworks, Columbus, OH, USA. Electron paramagnetic resonance (EPR) is an established and powerful tool for studying atomic-scale biomolecular structure and dynamics. Yet it requires a homogenous sample size of ~10^15 spin labeled biomolecules. Single molecule measurements provide improved insights into heterogeneous behaviors that can be masked by ensemble measurements and are often essential for illuminating the molecular mechanisms behind the function of a biomolecule. We report EPR measurements of single labeled biomolecule demonstrating the merging of these two powerful techniques. We selectively labelled individual double-stranded DNA molecules with nanodiamonds containing nitrogenvacancy (NV) centers, and optically detected the paramagnetic resonance of the single NV nanodiamond probe. Analysis of the spectrum reveals that the characteristic time scale for reorientation of the labeled molecule relative to the applied magnetic field is slow compared to the inverse of the EPR linewidth. This demonstration of EPR spectroscopic determination of the dynamics 337a of an individual labeled biomolecular construct provides the foundation for single molecule magnetic resonance studies of complex biomolecular systems. 1698-Plat Dynamics of Polymeric Protein Assemblies in Live Cells Revealed by Fluorescence Polarization Imaging of Single Molecules Shalin B. Mehta1, Molly McQuilken2, Patricia Occhipinti2, Amitabh Verma1, Rudolf Oldenbourg1, Amy S. Gladfelter2, Tomomi Tani1. 1 Marine Biological Laboratory, Woods Hole, MA, USA, 2Department of Biological Sciences, Dartmouth College, Hanover, NH, USA. Fluorescence single molecule imaging is a powerful means of investigating the distribution and dynamics of individual biological molecules and their assemblies. Molecular orientation underpins the biophysical properties of the biopolymers (e.g., DNA, cytoskeleton, extracellular matrix and misfolded protein oligomers). Therefore, methods that reveal the molecular orientation have yielded new insights in the structure and function of biomolecular assemblies. However, current methods are limited to measurement of orientation of single fluorophores and have not permitted robust imaging of orientation of many single molecules in parallel. We have developed a new fluorescence polarization microscope that instantaneously and efficiently sorts the emitted fluorescence along four polarization orientations (separated by 45 degrees) to provide instantaneous imaging of position and orientation of single fluorescent molecules and their assemblies. With aid of computational algorithms, the microscope provides diffraction limited spatial resolution, 10fps temporal resolution, and 10 degree of angular resolution in living cells. Our technique has enabled analysis of molecular position and orientation in vitro and in living cells. We found that phalloidin-Alexa Fluor 488 reports local orientation of sparsely labeled actin filaments. Taking advantage of this signal, we have measured changes in the orientation of local actin filaments as they undergo retrograde flow at the leading edge of migrating human keratinocytes. We also used our system to study organization of septins, a highly conserved cytoskeleton critical for cytokinesis and intracellular compartmentalization. We found that individual septin molecules labeled with constrained GFP attach to the cell cortex with consistent orientation. The consistent alignment of single septin-GFP suggested the presence of highly ordered scaffold. Our single molecule fluorescence orientation imaging technique is also promising to explore conformational changes of single molecules or mechanisms of protein assembly in living cells. 1699-Plat Single-Molecule-Sensitive FRET in Freely-Diffusing Attoliter Droplets Peker Milas, Sheema Rahmanseresht, Kieran P. Ramos, Ben D. Gamari, Lori S. Goldner. Physics, University of Massachusetts, Amherst, MA, USA. Fluorescence resonance energy transfer (FRET) from RNA confined in freelydiffusing attoliter aqueous droplets shows dramatic differences from that of RNA in solution. First, the use of droplet confinement is shown to substantially increase the signal-to-noise ratio of single-molecule sensitive measurements. However, a distinct shift in FRET is also observed. These differences can be attributed to a modified pH in the confining environment, which is a result of the well-documented autolysis of water and accumulation of hydroxide ions near the water interface. This outcome has implications for the use of droplets for protein crystallization, nanoparticle synthesis, biomicrofluidics, and analytical chemistry, where careful attention to the use of appropriate buffers or surfactants to control pH in the confined phase will be required. This work was funded by NSF MCB-0920139 and NSF DBI-1152386. Platform: Skeletal Muscle Mechanics, Structure, and Regulation 1700-Plat Activation and Relaxation Kinetics in Skeletal and Cardiac Muscles Srboljub M. Mijailovich1, Boban Stojanovic2, Djordje Nedic3, Michael A. Geeves4. 1 Chemistry and Chemical Bilogy, Northeastern Iniversity, Boston, MA, USA, 22Department of Mathematics and Informatics, University of Kragujevac, Faculty of Science, Kragujervac, Serbia, 3Department of Mathematics and Informatics, University of Kragujevac, Faculty of Science, Kragujervac, Serbia, 4School of Biosciences, University of Kent, Canterbury, United Kingdom. A large amount of data is available on the Ca2þ regulation of the thin filament of skeletal and cardiac muscle, but some general concepts are still under debate. To quantitatively describe muscle contraction and relaxation in the 3D multi-sarcomere geometry we have implemented in computational platform MUSICO: (i) a nine state actomyosin ATPase cycle, (ii) extensibility of thick 338a Tuesday, February 10, 2015 and thin filaments, (iii) the kinetics of Ca2þ binding to TnC involving two (skeletal) or one (cardiac) calcium binding sites and (iv) the McKillop-Geeves and the continuous-flexible-chain models of thin filament regulation. Simulated force-pCa relations closely follow the observations in skeletal and cardiac muscles. Interestingly, the higher Hill coefficient observed in cardiac muscle requires a longer confined persistence length (stiffer Tm) or a weaker interaction of Tm with actin. Note that confined persistence length increases with increase of Tm stiffness and decreases with strength of interaction of Tm with actin. The simulations also predicted delayed rapid muscle relaxation after a decrease of calcium concentration to pre- contraction level. The relaxation delay time was much shorter in cardiac muscle than in skeletal muscle. This is due to lower level of maximal activation of cardiac muscle compared to skeletal muscle, and therefore fewer TmTn units with more than one myosin bound. But the cardiac muscle takes a longer time to relax completely. These differences can be clearly seen in a comparison of cardiac and skeletal muscle twitch contractions. Using the same parameters, the simulations predicted the rise of force during the twitch but relaxation was slower than predicted by simulations, especially in cardiac muscle. This discrepancy invites further investigation of the interplay between the actomyosin cycle and calcium regulation via TmTn chain interactions with actin. Supported by: NIH R01s AR048776 and DC 011528 1701-Plat Alternative Versions of the Myosin Converter Vary Cross-Bridge Stiffness and Muscle Force Generation Douglas M. Swank, Bernadette M. Glasheen, Seemanti Ramanath, Qian Wang. Biological Sciences & Center for Biotechnology and Interdisciplinary Studies, Rensselaer Polytechnic Institute, Troy, NY, USA. The converter has been proposed to be the myosin elastic element that sets cross-bridge stiffness. According to most cross-bridge models, the elastic element’s stiffness should directly influence muscle force generation. When the elastic element is stretched by the power stroke, a stiffer element should increase force generation. We tested these hypotheses by using the natural variation in converter regions found in different Drosophila muscle types. In Drosophila, myosin isoforms are generated by alternative splicing from a single myosin heavy chain gene. The converter region is encoded by five alternative versions of exon 11. We modified the gene to express alternative versions in Drosophila indirect flight muscle (IFM) fibers instead of the wild-type 11a version. Using 0.125% muscle length sinusoidal oscillations at 500 Hz, we found that two of the four alternative versions, 11b (found in larval body wall muscles) and 11c (jump muscle) significantly decreased fiber rigor stiffness by 28% and 20%, respectively, indicating a decrease in cross-bridge stiffness. The changes in rigor stiffness (elastic modulus) roughly correlated (r2 ¼ 0.57) with force production by the fibers because 11b and 11c fibers also produced the lowest isometric tension. The correlation improved (r2 ¼ 0.71) when it was corrected for the influence of cross-bridge duty ratio on isometric tension. Relative duty ratio was estimated from the frequency at which maximum power was generated (fmax), and the muscle apparent rate constant 2pc. Converters 11b, 11d, and 11e decreased fmax by 26%, 34% and 43%, respectively. Thus, we conclude that the converter is at least part of the elastic element that sets cross-bridge stiffness, and that this element helps set natural variation in force production between muscle types. Supported by NIH grant R01 AR055611 to D.M.S. 1702-Plat Intermolecular Cooperativity of Skeletal Myosins Enhances Force Output in Myofilaments Motoshi Kaya1, Yoshiaki Tani2, Takumi Washio3, Toshiaki Hisada3, Hideo Higuchi2. 1 Physics, University of Calgary, Tokyo, Japan, 2Physics, University of Tokyo, Tokyo, Japan, 3Frontier Science, University of Tokyo, Tokyo, Japan. Muscle contraction is powered by the cyclic interaction of skeletal myosin molecules with actin filaments. Recent experiments suggest cooperative actions between myosin molecules, when part of an ensemble. In order to gain more insight into the mechanism of intermolecular cooperativity between myosins in a myofilament, displacements of actin generated by ~17 interacting myosins embedded in myosin-rod cofilament were measured by optical tweezers. Results showed stepwise displacements of actin (3-6 nm) under high loads of ~30 pN at every 1-2 ms. Dwell times gradually increased with increasing loads, which is distinctively different from highly load-dependent characteristics of dwell time observed in single myosins, implying that the number of force generating myosins increases with increasing loads. These results suggest that stepwise displacements may be generated by synchronous force generations of myosin molecules. In order to elucidate potential mechanisms of syn- chronous actions between myosins, we developed the simulation model, consisting of 17 myosins arranged in series with six transition states defined during actomyosin mechanochemical cycles. We have two types of model, implemented with either one or two power strokes. Interestingly, two power stroke model generates stepwise movements of actin at loads up to 30 pN, but one power stroke model generates forces up to 20 pN. These differences in force output are attributed to modulation of number of synchronous power stroke motors. The numbers of synchronous power stroke motors increase with increasing loads from 1.5 to 3 molecules in two power stroke model on average, while they are virtually the same in one power stroke model. Combined with results from both in-vitro and in-silico experiments, multiple power stroke states associated with strain-dependent kinetics are essential for synchronous force generations between myosins, that is a key feature for force enhancement of myosin ensembles. 1703-Plat Shortening-Induced Force Depression in Single Sarcomeres is Abolished by MgADP-Activation Neal Trecarten, Fabio C. Minozzo, Felipe S. Leite, Dilson E. Rassier. Kinesiology and Physical Education, McGill University, Montreal, QC, Canada. The mechanisms behind the shortening-induced force depression (FD) commonly observed in skeletal muscles remain unclear, but have been associated with sarcomere length non-uniformity and/or cross-bridge inhibition. The purpose of this study was twofold: (i) to evaluate if FD is present in isolated single sarcomeres, a preparation that eliminates sarcomere length nonuniformities and (ii) to evaluate if FD is inhibited when single sarcomeres are activated with MgADP, which biases cross-bridges into a strongly-bound state. Single sarcomeres (n¼16) were isolated from rabbit psoas myofibrils using two micro-needles (one compliant, one rigid), piercing the sarcomere externally adjacent to the Z-lines. The sarcomeres were contracted isometrically and subsequently shortened, in both Ca2þ- and MgADP-activating solutions. Shortening in Ca2þ-activated samples resulted in a 27.44% 5 9.04% force depression when compared to isometric contractions produced at similar final sarcomere lengths (P > 0.001). There was no FD in MgADP-activated sarcomeres (FD ¼ 1.79% 5 9.69%, P ¼ 0.435). These results suggest that FD is a sarcomeric property, and that is associated with an inhibition of myosin-actin interactions. 1704-Plat Force-Sarcomere Length Relations in Patients with Thin Filament Myopathy Caused by Mutations in NEB, ACTA1, TPM2, TPM3, KBTBD13, KLHL40 and KLHL41 Barbara Joureau1, J.M de Winter1,2, Christopher T. Pappas3, Edoardo Malfatti4, Alan Beggs5, Nigel Clarke6, Norma Romero7, Carol Gregorio3, Henk Granzier2, Ger J.M. Stienen8, Coen C.A Ottenheijm1,2. 1 VU University Medical Center , ICaR-VU, Dept. of Physiology, Amsterdam, Netherlands, 2University of Arizona, Dept. of Physiology, Tucson, AZ, USA, 3University of Arizona, Dept. of Cell Biology, Tucson, AZ, USA, 4Institut de Myologie, Groupe Hospitalier-Universitaire La Pitie´Salpeˆtrie`re, Paris, France, 5The Manton Center for Orphan Disease Research, Boston Children’s Hospital, Harvard Medical School, Division of Genetics and Program in Genomics, Boston, MA, USA, 6Institute for Neuroscience and Muscle Research, Children’s Hospital at Westmead, University of Sydney, Australia, 7Institut de Myologie, Groupe Hospitalier-Universitaire La Pitie´-Salpeˆtrie`re, AP-HP, UPMC-Paris6 UR76, INSERM UMR974, CNRS UMR 7215, Paris, France, 8VU University, Dept. of Physics and Astronomy, Amsterdam, Netherlands. Background: Mutations in NEB, ACTA1, TPM2, TPM3, KBTBD13, KLHL40 and 41 lead to thin filament myopathies, such as nemaline myopathy, congenital fiber type disproportion and cap disease. A hallmark feature of these myopathies is muscle weakness. Here, we aimed to elucidate whether mutations in NEB, ACTA1, TPM2, TPM3, KBTBD13, KLHL40 and KLHL41 affect the maximal force generating capacity of muscle fibers. Subsequently, by determining the sarcomere length-dependence of force, we investigated whether changes in thin filament length contributed to muscle weakness. Methods: Biopsies from NM, CFTD, and CAP patients (n¼39) with mutations in NEB, ACTA1, TPM2, TPM3, KBTBD13, KLHL40 or KLHL41 were compared to biopsies from healthy controls (n¼9). Using permeabilized muscle fibers, maximal active tension was determined at incremental sarcomere lengths (range 2.0-3.5 mm) to obtain the force-sarcomere length relationship. Results: The maximal active tension (Fmax (in mN/mm2, mean5SEM)) was significantly lower in biopsies from NEB (4655), ACTA1 (4854), TPM2 (7158), TPM3 (85510), KBTBD13 (7853), KLHL40 (2.850.2) and KLHL41 (6354) patients compared to biopsies of controls (12957). Tuesday, February 10, 2015 No shift in the force-sarcomere length relationship was observed in TPM3, TPM2, KBTBD 13, KLHL 40 and KLHL41 patients. In contrast, several patients with ACTA1 and NEB mutations showed a leftward shift of the forcesarcomere length relationship indicating shorter thin filaments. Furthermore, the slope of the descending limb of the force-sarcomere length relationship in these patients is less steep than the slope of the CTRL curve, suggesting heterogeneity of thin filaments. Conclusion: Our data suggest that mutations in NEB and ACTA1 result in changes in thin filament length. Insights in the mechanisms underlying weakness in patients with thin filament mutations are necessary to improve specific treatment strategies. 1705-Plat An Active Role for the Z-Band during Contraction and Relaxation Lloyd Zhao, Lanette R. Fee, Sehyang Han, Michael K. Reedy, Robert J. Perz-Edwards. Cell Biology, Duke University, Durham, NC, USA. In striated muscle, the Z-band transmits force between contracting sarcomeres; however, it is not merely a static anchor for actin filaments cross-linked by alpha-actinin. Shimamoto observed a coordinated yielding of adjacent sarcomeres in response to stretch, and its disruption by an alpha-actinin antibody [1], indicating an active Z-band function and communication between adjacent sarcomeres. We hypothesized the inter-sarcomeric communication must involve a previously observed but poorly understood, reversible transition between two Z-band structures descriptively called small-square and basketweave [2], implying that alpha-actinin antibody must shift the equilibrium between the two states. Using EM, we imaged Z-bands of chemicallyskinned relaxed rabbit soleus fibers and found that alpha-actinin antibody induces complete conversion to the basketweave form of the Z-band, whereas control fibers without antibody show the expected small square form. Since the small-square form was previously associated with relaxed fibers and basketweave with contracted fibers [2], our results suggest that the basketweave Z-band may store elastic energy, which can then favor or drive sarcomere relaxation when the Z-band reverts to the small-square structure. By locking the Z-band in the basketweave form, the antibody prevents the relaxation and concomitant yielding of adjacent sarcomeres previously observed [1]. In parallel mechanics experiments, we found antibody treatment did not interfere with maximal calcium-activated isometric contraction and relaxation; however isometric force and stretch-activation force during sub-maximal activation by ADP were both reduced after antibody treatment. Thus, the alteration of Z-band form by antibody only affects the muscle’s dynamic response to stretch during partial activation. 1. Shimamoto, Y, et al. Inter-sarcomere coordination in muscle revealed through individual sarcomere response to quick stretch. PNAS, 2009. 106(29):11954-9. 2. Goldstein, MA, et al. The Z-band lattice in skeletal muscle before, during and after tetanic contraction. J Muscle Res, 1986. 7(6):527-36. 1706-Plat Effects of Cardiac Myosin Binding Protein-C on Actin Motility are Explained with a Drag-Activation-Competition Model Sam Walcott1, Steffen Docken1, Samantha P. Harris2. 1 Mathematics, University of California, Davis, Davis, CA, USA, 2Cellular and Molecular Medicine, University of Arizona, Tucson, AZ, USA. At the molecular level, muscle contraction is powered by cyclic interactions between the molecular motor myosin and actin filaments. In vertebrate striated muscle, this interaction is regulated by calcium binding to troponin and a resulting displacement of tropomyosin that renders binding sites on actin accessible to myosin. Other proteins are involved in this process, but their roles are not as well understood. One such protein is myosin binding protein-C (MyBP-C). The cardiac isoform of this protein (cMyBP-C) has attracted recent attention because of its role in heart disease. Despite this interest, its function is not well understood, partly because in some assays it can have multiple effects. In the actin motility assay, for example, cMyBP-C can have a dual, biphasic activating/inhibitory effect. To elucidate the function of cMyBP-C, we developed a mathematical model of its interaction with the contractile proteins actin and myosin as well as the regulatory protein tropomyosin. This is, to our knowledge, the first mechanistic model of cMyBP-C’s function. We used this model to fit published measurements of cMyBP-C in the actin motility assay. These fits demonstrate that a drag-activation-competition mechanism is consistent with the data, while models lacking either drag or competition are not. These three effects can arise simply from cMyBP-C binding to actin: cMyBP-C binding to actin both displaces tropomyosin (leading to activation) and also precludes concurrent myosin binding (leading to competition). Additionally, if cMyBP-C is attached 339a to the flow cell surface, then binding to actin forms a transient link between the actin filament and the surface, generating a viscous drag that further slows motility. Future use of this model may help distinguish mechanistic effects of cMyBP-C mutations that affect actin binding. 1707-Plat The Myosin Super-Relaxed State is Regulated by Estradiol Brett A. Colson1, Karl J. Petersen1, Brittany C. Collins2, David D. Thomas1, Dawn A. Lowe2. 1 Biochem, Molec Biol, and Biophysics, University of Minnesota, Minneapolis, MN, USA, 2Physical Medicine and Rehabilitation, University of Minnesota, Minneapolis, MN, USA. We have used quantitative epifluorescence microscopy of fluorescent ATP to measure single-nucleotide turnovers in skinned muscle fibers from mouse models of female aging and hormone treatment. The loss of muscle strength is an undesirable consequence of aging, often leading to frailty, disability, and loss of independence for the elderly. Female muscle is additionally affected by age due to reduction of ovarian hormone production with menopause. Estradiol (E2) is the key hormonal signal to skeletal muscle in females, and strength loss is attenuated by E2 treatment. Single skeletal muscle fibers, isolated from sham-operated or ovariectomized (OVX) mice with or without E2 treatment, were incubated with mantATP. We measured decay of mantATP fluorescence intensity in an ATP chase experiment, as pioneered by Cooke and colleagues (Stewart et al., 2010; Cooke, 2011). These studies by Cooke unveiled a novel regulated state of muscle myosin characterized by slow nucleotide turnover on the order of minutes, termed the super-relaxed state (SRX). We detected a slow phase of nucleotide turnover in approximately one-third of sham-operated myosin heads, consistent with SRX. Turnover was substantially faster in OVX fibers. Strikingly, the chronic lack of E2 in OVX fibers did not alter the fraction of heads with slow turnover but rather decreased the turnover time, suggesting disordering of SRX. E2 treatment in OVX mice partially reversed this effect on SRX, while acute E2 treatment in the muscle bath had no effect. All experiments were conducted with uniformly low myosin light chain phosphorylation. We conclude that E2-mediated signaling regulates slow ATP turnover and ordering of heads, probably via pathways distinct from myosin phosphorylation status. Age- and hormone-related muscle functional losses may be targetable at the level of thick filament structure for strategies to offset weakness and metabolic changes that occur with age. Platform: Intracellular Channels and Calcium Sparks and Waves 1708-Plat Structural Insights into the Nature of the Unique Anion Binding Site within the Cardiac Ryanodine Receptor N-Terminal Region and Associated Disease Mutations Siobhan Wong1, Michele Bedin2, Filip Van Petegem1. 1 University of British Columbia, Vancouver, BC, Canada, 2University of Gothenburg, Gothenburg, Sweden. Ryanodine receptors (RyRs) are large intracellular calcium-release channels of the endo/sarcoplasmic reticulum that play a critical role in mediating excitation-contraction coupling. Mutations within the skeletal muscle isoform (RyR1) are associated with the pharmacogenetic disorder known as Malignant Hyperthermia (MH) as well as congenital myopathies. Concurrently, mutations within the cardiac (RyR2) isoform have been linked to life-threatening arrhythmias, such as catecholaminergic polymorphic ventricular tachycardia (CPVT), a potential precursor of sudden cardiac death. While recent high-resolution structures have provided some mechanistic insights as to how mutations may impact receptor function, this subject requires more work. The RyR1 aminoterminal region contains an elaborate ionic interaction network embedded between its three domains which is key to the protein’s stability. Despite all the ionic interaction partners being conserved within RyR2, the replacement of two histidine residues with Tyr125 and Arg420 within RyR2 is enough to abolish the ionic network and introduce a unique central anion binding site within RyR2. Here, we present structural insights into the nature of the ˚ crystal structure has been solved of the RyR2 anion binding site. A 2.6A amino-terminal RyR2 region (residues 1-547) into which two histidines, Y125H and R420H, were introduced which abolished chloride binding but which failed to restore the ionic network of its counterpart in RyR1. The amino-terminal intrasubunit domain-domain interfaces of both RyR1 and RyR2 are each targeted by over twenty disease-associated mutations. Here, we provide the biochemical and structural characterization of a novel MH RyR1 mutation, H113Q, and a potential CPVT-causing mutant, RyR2 R420W, both of which reside at a domain-domain interface. 340a Tuesday, February 10, 2015 1709-Plat Crosstalk between RyR2 Oxidation and Phosphorylation Contributes to Cardiomyopathy in Mice with Duchenne Muscular Dystrophy George G. Rodney1, Qiongling Wang1, Guoliang Wang1, Xander H.T. Wehrens1,2. 1 Molecular Physiology & Biophysics, Cardiovascular Research Institute, Baylor College of Medicine, Houston, TX, USA, 2Department of Medicine/ Cardiology, Baylor College of Medicine, Houton, TX, USA. Patients with Duchenne muscular dystrophy (DMD) are at risk of developing cardiomyopathy and cardiac arrhythmias. Studies in a mouse model of DMD revealed that enhanced sarcoplasmic reticulum (SR) Ca2þ leak contributes to the pathogenesis of cardiac dysfunction. In view of recent data suggesting the involvement of altered phosphorylation and oxidation of the cardiac ryanodine receptor (RyR2)/ Ca2þ release channel, we hypothesized that inhibition of RyR2 phosphorylation in a mouse model of DMD can prevent SR Ca2þ leak by reducing RyR2 oxidation. Confocal Ca2þ imaging and single RyR2 channel recordings revealed that inhibition of either S2808 or S2814 phosphorylation, or inhibition of oxidation could normalize RyR2 activity in mdx mice. Moreover, genetic inhibition of RyR2 phosphorylation at S2808 or S2814 reduced RyR2 oxidation. Production of reactive oxygen species (ROS) in myocytes from mdx mice was reduced by both inhibition of RyR2 phosphorylation or the ROS scavenger 2-mercaptoproppionylglycin (MPG). Finally, it was shown that ROS production in mdx mice is proportional to the activity of RyR2-mediated SR Ca2þ leak. We conclude that increased reactive oxygen species (ROS) production in the hearts of mdx mice drives the progression of cardiomyopathy. Inhibition of RyR2 phosphorylation can suppress SR Ca2þ leak in mdx mouse hearts in part by reducing RyR2 oxidation. 1710-Plat Secretoneurin, a Novel Endogenous CaMKII Inhibitor, Augments Cardiomyocyte Calcium Handling and Inhibits Arrhythmogenic Calcium Release Anett H. Ottesen1, Cathrine R. Carlson2, Andrew G. Edwards2, Ole J.B. Landsverk3, Rune F. Johansen4, Morten K. Moe5, Magnar Bjøra˚s4, Mats Stridsberg6, Torbjørn Omland1, Geir Christensen2, Helge Røsjø1, William E. Louch2. 1 Division of Medicine, Akershus University Hospital, Lørenskog, Norway, 2 Institute for Experimental Medical Research, Oslo University Hospital Ulleva˚l, Oslo, Norway, 3Department of Pathology, Oslo University Hospital Rikshospitalet, Oslo, Norway, 4Department of Microbiology, Oslo University Hospital Rikshospitalet, Oslo, Norway, 5Division of Diagnostics and Technology, Akershus University Hospital, Lørenskog, Norway, 6 Department of Medical Sciences, Uppsala University, Uppsala, Sweden. Secretoneurin (SN) is the functional fragment of secretogranin II, a granin protein that is increased in heart failure patients and associated with mortality. In non-cardiac cells, SN has been shown to alter Ca2þ homeostasis. We presently investigated the effects of exogenous SN treatment on Ca2þ handling in heart. SN was observed to be rapidly taken up into intact hearts, and internalized into cardiomyocytes via endocytosis. Bioinformatic analyses suggested potential interactions between SN and calmodulin (CaM), and also the catalytic region of Ca2þ/CaM-dependent protein kinase II d (CaMKIId). These putative interaction sites were confirmed by employing pull-down, immunoprecipitation, and Biacore analyses. SN attenuated CaMKIId activity in a dose-dependent manner, and reduced autophosphorylation of CaMKIId in Langendorff-perfused hearts, both in the absence and presence of isoproterenol. SN also reduced basal and isoproterenol-induced CaMKIIddependent ryanodine receptor phosphorylation, and CaMKIId-dependent phosphorylation of phospholamban. In isolated cardiomyocytes, exogenous SN (2.8 mmol/L) increased the magnitude and kinetics of cardiomyocyte Ca2þ transients and contractions. In agreement with reduced CaMKIIddependent ryanodine receptor phosphorylation, Ca2þ spark frequency and dimensions were reduced, and sarcoplasmic reticulum Ca2þ content was increased. Augmentation of Ca2þ transients occurred despite reduction in the magnitude of L-type Ca2þ current, as expected following CaMKIId inhibition. During challenge with isoproterenol, SN treatment reduced the frequency of arrhythmogenic Ca2þ waves. Similarly, in patch-clamped cells stimulated with action potentials, the appearance of delayed afterdepolarizations and spontaneous beats was inhibited by SN. In conclusion, SN is a novel CaMKIId inhibitor that inhibits ryanodine receptor Ca2þ leak, resulting in augmented sarcoplasmic reticulum Ca2þ content, Ca2þ transients, and contractions with reduced occurrence of arrhythmogenic Ca2þ waves and delayed afterdepolarizations. These findings suggest that increased SN levels during heart failure may be compensatory in the most severely ill patients. 1711-Plat Large-Scale, Automated Calcium Spark Analysis using iSpark Reveals Functional and Spatial Remodeling During Cardiac Hypertrophy Qinghai Tian1, Laura Schro¨der1, Aline Flockerzi1, Andre Zeug2, Lars Kaestner1, Peter Lipp1. 1 Institute for Molecular Cell Biology, Saarland University, Homburg/Saar, Germany, 2Institute for Cellular Neurophysiology, Medizinische Hochschule Hannover, Homburg/Saar, Germany. Understanding of cardiac Ca signaling is driven by advancements in imaging and analysis tools. The availability of fast line scanning allowed the identification of Ca sparks that revolutionized our appreciation of cardiac Ca signaling in the healthy and diseased heart. Ultra-high-speed confocal imaging of three dimensional (2D over time) Ca spark properties yields novel high content data about such signals whose characteristics eventually determine the performance of the entire heart. The full potential of such data remains concealed owing to the lack of comprehensive, fully automated and unbiased analysis tools. We have developed an intelligent software tool (iSpark) employing fully automatic, self-adaptive and unbiased algorithms to investigate Ca sparks in cardiac myocytes from healthy animals and mice with tiered stages of hypertrophy. Long-term recording of Ca sparks with high speed 2D over time confocal imaging of permeabilized ventricular myocytes produced high content spark data. With iSpark we explored 670,000 individual events and revealed that their subcellular arrangement, amplitude and frequency were substantially altered with the severity of cardiac hypertrophy. While line scanning of our data failed to show any correlation, iSpark-based analysis demonstrated a malfunctional microscopic Ca signaling strongly correlating with the disorganization of T-tubules and the severity of hypertrophy. The availability of highly sophisticated analysis tools, such as iSpark, substantially fosters large-scale data exploration. iSpark-based analysis of large-scale Ca spark data revealed a causal contribution of distinct subcellular remodeling of Ca handling to the progression of cardiac hypertrophy. 1712-Plat Regulation of Calcium Clock-Mediated Pacemaking by Inositol-1,4,5Trisphosphate Receptors in Mouse Sinoatrial Nodal Cells Nidhi Kapoor1, Andrew Tran1, Jeanney Kang1, Rui Zhang1, Kenneth D. Philipson2, Joshua I. Goldhaber1. 1 Heart Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA, 2 Department of Physiology, University of California, Los Angeles, CA, USA. Sinoatrial node (SAN) automaticity is attributable to the interplay of several membrane currents such as funny current (If) and the Na-Ca exchanger (NCX) current activated in response to ryanodine receptor (RyR) mediated Ca release from the sarcoplasmic reticulum (SR). Whether another SR Ca release channel, the inositol-1,4,5-triphosphate receptor (IP3R), can influence automaticity in SAN is controversial, in part due to the confounding influence of periodic Ca flux through the sarcolemma accompanying each beat. We used atrial-specific NCX knockout (KO) SAN cells to study IP3 signaling in a system where periodic [Ca]i cycling persists despite the absence of depolarization or Ca flux across the sarcolemma. We recorded confocal line scans of spontaneous Ca release in WT and NCX KO SAN cells, in the presence or absence of an IP3R blocker (2-APB) or during inhibition of phospholipase C by U73122. We found that superfusion with 2APB (2 mM) decreased the frequency of Ca transients in WT by 82.7% (n¼9, p<0.05) and Ca waves in NCX KO by 69.9% (n¼10, p<0.05). Similar results were also obtained with U73122 (1 mM). Alternatively, increased IP3 production induced by phenylephrine (PE; 10 mM) increased Ca transient frequency in WT (n¼8. P> 0.05) and Ca wave frequency in KO cells (n¼9, p <0.05) that was reversed by 2-APB. To determine if IP3Rs exert their modulatory effect on pacemaking via RyRs, we recorded Ca transients during application of PE in the continued presence of ryanodine at a blocking concentration (100 mm) that does not deplete SR stores. Under these conditions PE was unable to restore Ca transients. Thus, Ca release from IP3Rs can modulate the ‘‘Ca clock’’ processes that regulate pacemaker frequency in the murine SAN via Ca-induced Ca release through the RyRs. 1713-Plat Structural Studies of IP3R by Cryoem Qiu-Xing Jiang, Hui Zheng, Marc Llaguno. Cell Biology MC9039, UT Southwestern Medical Center, Dallas, TX, USA. Inositol 1,4,5-trisphosphate receptors (IP3Rs) play important roles in a battery of cellular activities. Structural study of the receptors is therefore very important for understanding how they are gated by their natural ligands and are modulated by their intracellular partners. In the past several years, multiple groups have generated very disparate reconstructions of the type 1 IP3R. Tuesday, February 10, 2015 Striking structural variations have been reported for the receptors in different detergents and for the receptors prepared from native and sf9 cells by the same research groups, suggesting that biochemical preparations of the receptors might have had significant variations and that the heterogeneity in the samples could be a limiting factor in reaching accordant results. To resolve such discrepancies, we developed a three- layered strategy to enhance biochemical homogeneity and structural agreements. We collected two cryoEM datasets of the receptors in the absence and presence of IP3 and Ca2þ and calculated two separate reconstructions. The two structures not only agree with each other in many aspects, but also reveal a conformational change at the top of the cytosolic domain that may lead to some reorganization of the channel pore. In order to verify the structural details and solidify the conformational changes in the pore domain, we need higher resolution structures. We are using the highend facilities with direct electron detectors to collect near-atomic resolution data in order to further improve the resolutions of our 3D reconstructions. 1714-Plat Multiple Closed States of the Ryanodine Receptor Determined by CRYOEM Pablo Castro-Hartmann, Joshua Lobo, Montserrat Samso. Physiology and Biophysics, VCU School of Medicine, Richmond, VA, USA. Ryanodine receptors (RyRs) are intracellular ion channels involved in Ca2þ release from internal stores in excitable cells. These channels are the largest channels known and are homotetramers, sizing ~2,26 MDa. The 3D structure of RyR1 in it open and close states was determined previously, revealing that the ion gate opening mechanism rely on long-range conformational ˚ . The RyR gating properties are highly regulated by changes over 100 A Ca2þ, Mg2þ, ATP, and FKBP12. The native conformation of RyR1 in presence of physiological concentrations of Mg2þ and ATP is unknown. Here we determine the 3D structure of RyR1 in non-activating conditions (submicromolar Ca2þ) in the presence of Mg2þ and an ATP analog, but in a flexible conformation by absence of FKPBP12. This new structure was determined using cryoEM and image processing. The resulting 3D structure is in the closed conformation when compared to 3D reconstructions of RyR1 in open and closed conditions in presence of FKBP12 determined previously. In addition, from the comparison among several 3D reconstructions, we establish new conformation-function correlations. We find that the rhomboid structures formed by domains 2-4-5-6 situated far away from the ion gate move as a whole during gating, and define a ‘‘flexion angle’’ that appears to be correlated with the degree of opening of the channel, whereby the flexion angle after adding Mg2þ and ATP shifts by 3 degrees towards the closed state. In conclusion this research suggests that physiological concentrations of Mg2þ and ATP shift the RyR1 conformation toward the closed conformation and also suggests that the closed conformation encompasses sub-states. 1715-Plat Crystal Structures of the Ryanodine Receptor SPRY2 Domain Kelvin Lau, Filip Van Petegem. Life Sciences Institute, Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC, Canada. The SPRY2 domain is one of three repeats of the same fold that are present within the RyR. It has been suggested as a key protein interaction site with dihydropyridine receptors to mediate excitation-contraction coupling in skeletal muscle tissue. RyR1 and RyR2 SPRY2 domains were crystallized and reveal differences with what was thought to be SPRY2 and with several other known SPRY domain structures. Our RyR1 SPRY2 construct (rabbit, 1070-1246) is 43% larger than a previously reported construct (1085-1208), consists of multiple modules, is highly soluble, and is monomeric in nature. Docking of the RyR1 SPRY2 structure places it in between the central rim and the clamp region. The structure of RyR2 SPRY2 (mouse, 1080-1253) and a loss-offunction disease mutant (human, T1107M) causing cardiomyopathy were also determined. The T1107M mutation is buried and causes a large ~10 C decrease in thermal stability as compared to wildtype and also causes local misfolding of the domain. Finally, RyR1 SPRY2 binding to the DHPR II-III loop is undetectable by isothermal titration calorimetry. Platform: Membrane Structure 1716-Plat Interaction of HIV-1 Gag Protein’s Ma Membrane Binding Domain with Membrane Mimics Probed by Low- and Wide-Angle X-Ray Scattering Lauren O’Neil1, Leah Langer1, Davina Perera2, Zachary Dell1, John F. Nagle1, Stephanie Tristram-Nagle1. 1 Physics Dept., Carnegie Mellon University, Pittsburgh, PA, USA, 2 Biomedical Engineering, Douglass College, Rutgers University, NJ, USA. 341a The structural Gag protein from the HIV-1 virus is required for assembling new virus particles within an infected T-cell; Gag’s binding to the inner leaflet of the host plasma membrane is the first step in this process. In an effort to understand the molecular and energetic details of this binding, we studied the N-terminal 31 amino acids of Gag’s MA membrane binding domain with lipid membrane mimics: PC, PC:PE, PC:PS, PC:PI, PC:PIP, PC:PIP2, PC:PE:PI, PC:PE:PIP, PC:PE:PIP2, PC:PE:PIP2:cholesterol and PC:PE:PS:cholesterol in various molar ratios. Oriented, fully hydrated lipid mimics with/without the myristoylated (MAmyr) and non-myristolyated (MA) peptide were X-rayed at the Cornell High Energy Synchrotron Source. We found that MAmyr lowered KC (softened) pure POPC membranes more than did MA; in general, both peptides lowered KC for all 18 mimics. MAmyr increased slightly Sxray (chain order) of PS-containing membrane mimics, but both peptides decreased Sxray slightly with increasing concentrations of PI, PIP or PIP2 when mixed with POPC. When POPC:POPE was mixed with PI, PIP or PIP2 (5:3:2), PI decreased, PIP had no effect and PIP2 increased chain order with either peptide compared to controls. The head-to-head spacing (DHH) was decreased by both peptides ˚ ) in most mimics. In pure POPC, or when PI, PIP or PIP2 was mixed (1-3 A with POPC, modeling suggested a penetration of MAmyr into the hydrocarbon region, compared to an interfacial headgroup position for MA. Therefore the effect of MA peptides on membrane structure and properties depends on the composition of the lipid mimics as well as on the myristoyl group. Acknowledgments: NIH R01 GM44976 (STN and JFN). 1717-Plat Influence of Domain Size on Structure and Elastic Fluctuations in Complex Lipid Mixtures Peter Heftberger1, Benjamin Kollmitzer1, Frederick Heberle2, Jonathan Nickels2, John Katsaras2, Georg Pabst1. 1 Institute of Molecular Biosciencies, Biophysics Division, NAWI Graz, BioTechMed-Graz, University of Graz, Graz, Austria, 2Neutron Sciences Directorate, Oak Ridge National Laboratory, Oak Ridge, TN, USA. Lipid-only domains are well-established mimetic systems for membrane rafts enabling the study of their physical properties under strictly controlled conditions. Of particular interest are four component lipid mixtures entailing the variation of lipid domain size from micron regime down to a few nanometers [1]. Applying our recently developed small-angle x-ray scattering data analysis technique, we have studied changes of membrane thickness, lateral lipid packing and bending fluctuations for coexisting liquid-ordered (Lo) and liquiddisordered (Ld) phases in DOPC/POPC/DSPC/cholesterol mixtures along this domain-size trajectory, including the melting of Lo domains as a function of temperature. Bending fluctuations for coexisting Lo domains were found to be significantly lower than for single Lo phases at the boundary of the LoþLd regime. In turn, little variation was observed when domains exceeded sizes of 160 nm. Further, we found that the melting of Lo domains as a function of temperature is controlled by thickness differences between Lo and Ld and the associated domain line tension. This work was supported by Austrian Science Fund FWF, Project No. P24459-B20. [1] F.A. Heberle et al., J. Am. Chem. Soc. 135 (18), 6853-6859 (2013) 1718-Plat Phase Coexistence in Lipid Membranes Induced by Buffering Agents and Charged Lipid Headgroups Merrell A. Johnson1, Soenke Seifert2, Millicent A. Firestone3, Horia I. Petrache1, Ann C. Kimble-Hill4. 1 Dept. of Physics, IUPUI, Indianapolis, IN, USA, 2X-Ray Science Division, Advanced Photon Source, Argonne National Laboratory, Argonne, IL, USA, 3 Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, NM, USA, 4Dept. of Biochemistry & Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, USA. Recent literature has shown that buffers affect the interaction between lipid bilayers through a mechanism that involves van der Waals forces, electrostatics, hydration forces and membrane bending rigidity. We endeavour to show phase coexistence as an effect of charges from the aqueous boundary layer on the mixed chain 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayers. We will present data that suggests one phase dehydrates below the value in pure water while the other phase swells as the concentration of buffer is increased. However, since the two phases must be in osmotic equilibrium with one another, this behavior challenges theoretical models of lipid interactions and introduces new variables to consider for the Gibbs phase rule. This model of lipid charging was then applied to explain the mechanisms behind phase separation in lipid mixtures containing phosphatidylinositols. This work was supported by NIH NCI K01-CA169078-01 and Indiana University Collaborative Research Grants. 342a Tuesday, February 10, 2015 1719-Plat Analysis of PI(4,5)P2 Lateral Organization at the Plasma Membrane of Living Cells Through FRET Maria Joa˜o Sarmento1, Ana Coutinho1,2, Manuel Prieto1, Fa´bio Fernandes1. 1 Centro de Quı´mica-Fı´sica Molecular and IN, Instituto Superior Te´cnico Univ. Lisbon, Lisbon, Portugal, 2Dep. Quı´mica e Bioquı´mica, Faculty of Sciences - Univ. Lisbon, Lisbon, Portugal. Phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) is an important component of the inner leaflet of the plasma membrane of eukaryotic cells. Despite the fact that it only comprises approximately 1 mol% of the total membrane phospholipids, this phosphoinosite has been associated with many different cells functions including membrane trafficking, actin cytoskeleton remodeling and cell motility, among others. The physiological functions of this lipid seem to depend on localized concentration fluctuations within the plasma membrane. In fact, the distribution of this lipid in the plasma membrane has been proposed to be heterogeneous, and PI(4,5)P2 clustering is detected on model membranes under specific conditions. Domains highly enriched in PI(4,5)P2 were also reported at the plasma membrane of specific cell types. However, for most cellular models, scarce evidence has been found for PI(4,5)P2 segregation/clustering in the plasma membrane. In this context, our main goal was to study the distribution of PI(4,5)P2 molecules in cells lines where no heterogeneity in PI(4,5)P2 lateral distribution had been previously observed. The distribution of PI(4,5)P2 was assessed from FRET microscopy measurements with pleckstrin homology (PH) domains tagged with different fluorescent proteins. We applied a FRET methodology capable of discriminating between FRET from aggregates/clusters from noninteracting molecules within the plasma membrane of living cells. Our results clearly show distinct PI(4,5)P2 local densities in different cellular models, suggesting different patterns of PI(4,5)P2 lateral distributions within the plasma membrane. In addition, the effect of cholesterol removal on PI(4,5)P2 lateral organization is significantly different in distinct cell lines, suggesting that the role of cholesterol in the formation of PI(4,5)P2 enriched domains varies considerably. This work was supported by FCT - Foundation of Science and Technology (PTDC/QUI-BIQ/119494/2010 and RECI/CTM-POL/0342/2012). M.J.S. and F.F. acknowledge research grants (SFRH/BD/80575/2011 and SFRH/BPD/ 64320/2009) from FCT. 1720-Plat Impact of PI(3,4,5)P3-Mediated Beta-Arrestin-1 Recruitment on Structure of Asymmetric Lipid Bilayers Achebe N.O. Nzulumike1, Signe Mathiasen1, Jacob P. Mahoney2, Marite´ Ca´rdenas Go´mez1, Dimitrios G. Stamou1, Kell Mortensen1. 1 Niels Bohr Institute, University of Copenhagen, Copenhagen, Denmark, 2 Department of Pharmacology, University of Michigan Medical School, Ann Arbor, MI, USA. The membrane spanning G-protein coupled receptors (GPCRs) facilitate crucial physiological responses to a variety of extracellular ligands, such as hormones, neurotransmitters, ions, photons, and other stimuli [1], thus 50% of drugs available in the market today target GPCRs [2]. The biological mechanisms behind GPCR-signaling involves several other biomolecules, including the key protein group of b-arrestins. These cytosolic adaptor proteins regulate signaling through several different pathways, classically by removing receptors from the plasma membrane via clathrin-mediated endocytosis [3]. In membrane binding experiments using both confocal fluorescence microscopy and quartz crystal microbalance, we found that b-arrestin-1 binds specifically to supported lipid bilayers (SLBs) containing phosphatidylinositol trisphosphate lipids, PI(3,4,5)P3. We have consistently shown, using both methods, that such binding is lipid specific and not driven by membrane charge. We have also shown that the membrane-protein interaction depends on protein concentration and membrane composition, and that b-arrestins can induce membrane curvature, which may play a role in the endocytic pathway. This is for the first time quantified with respect to dynamics and respective contributions of specific vs non-specific binding. Having established an assay for PI(3,4,5)P3-mediated recruitment of b-arrestin-1, we conducted a series of neutron reflection experiments to study the protein-lipid interaction on a structural level. Both hydrogenated and deuterated protein was used in reflectometry experiments with diverse contrast matching of the bulk media. We present here the detailed structures of asymmetric lipid bilayers containing PI(3,4,5)P3, and demonstrate that b-arrestin1 not only binds but also reorganizes the membrane structure. 1. Rosenbaum, D. M. et al. Nature. 459(7245), 356-363 (2009). 2. Gudermann, T. et al. J Mol Med-Jmm. 73(2), 51-63 (1995). 3. DeWire, S. M. et al. Annu Rev Physiol. 69, 483-510 (2007). 1721-Plat Computational Lipidomics and the Lipid Organization of Cell Envelopes Helgi I. Ingolfsson1, Manuel N. Melo1, Tsjerk A. Wassenaar1, Xavier Periole1, Alex H. de Vries1, D. Peter Tieleman2, Siewert J. Marrink1. 1 University of Groningen, Groningen, Netherlands, 2University of Calgary, Calgary, AB, Canada. The detailed lipid organization of cellular membranes remains elusive. A typical plasma membrane contains hundreds of different lipid species that are actively regulated by the cell. Currently over 30,000 biologically relevant lipids have been identified and specific organisms often synthesize thousands of different lipid types. This is far greater diversity than is needed to maintain bilayer barrier properties and to solvate membrane proteins. Why organisms go through the costly progress of creating and maintaining such a large diversity of lipids is one of the big open questions in biology. What is the individual role of these lipids, and how do they interact and organize in the membrane plane? To start to address these questions we optimized and developed the Martini coarse-grained force field lipidome. We systematically explore over a hundred different lipid types using the Martini model. Bulk properties of each individual lipid type (e.g. bilayer thickness, area per lipid, diffusion, order parameter and area compressibility) were analyzed and overall trends compared to experimental values. Using pre-existing Martini lipids and the newly characterized ones, idealized plasma membranes containing dozens of different lipid types asymmetrically distributed between the membrane leaflets were constructed and simulated on the multi microsecond time scale. In terms of lipid composition these are by an order of magnitude the most complex simulations to date. These large-scale simulations provide a high-resolution view on the lipid organization of plasma membranes at an unprecedented level of complexity and allow us to analyze a variety of plasma membrane physicochemical properties, including: lipid-lipid interactions, bilayer bulk material properties, domain formation and coupling between the bilayer leaflets. Overall, the plasma membranes show global non-ideal mixing of different lipid species at different spatiotemporal scales. 1722-Plat Phase Behavior of Synaptosomal Membranes: The Effect of Lipid Composition and Temperature Atsuko Kimura1, Gulcin Pekkurnaz2, Tomohiro Kimura1, Sierra C. Germeyan1, Jessica Zimmerberg-Helms1, James Loewke1, Paul S. Blank1, Thomas S. Reese1, Klaus Gawrisch1, Ludmila Bezrukov1, Joshua Zimmerberg1. 1 National Institutes of Health, Bethesda, MD, USA, 2F.M. Kirby Neurobiology Center, Boston Children’s Hospital, Boston, MA, USA. The phase behavior of synaptosomal membranes isolated from mammalian rat and marine invertebrate squid intact nerve endings was compared. Homogeneous liquid disordered (La) phase was observed at their body temperatures of 37 and ~20 C for the rat and squid, respectively, using fluorescence microscopy and 1H-MAS NMR. Temperature decrease resulted in co-existence of an ordered phase with onset temperatures of 24 C for rat and 8 C for squid. Comprehensive analyses of the lipid composition by LC/MS explained the significant difference in the onset temperature: >2x higher content of 18:0 and 18:1 lipid chains in rat, which contributes to a higher transition temperature. The amount of u-3 chains was ~3x higher in squid than in rat due to a difference in the amounts of 22:6 and 20:5: squid membranes contained 2x more total 22:6 in major phospholipids (PC, PE, and PS) and cardiolipin (17:0,22:6,22:6,22:6-species). While prominent in rat, 20:4 was not found in squid. It is known for La phase PC that 16:0,22:6 is more ordered and thicker with a smaller area/lipid than 16:0,20:4. The high content of 22:6 in squid may contribute to order in the La phase at the body temperature (~20 C) in contrast to the high content of 20:4 in rat at a higher body temperature (37 C). Fluorescence microscopy data showing the temperature-dependent phase behavior using giant unilamellar vesicles prepared with lipid compositions based on the synaptosomal measurements above will be discussed. Synaptosomal preparations of squid with body temperatures differing by ~9 C will be compared. 1723-Plat Morphology Induced Receptor Trapping in Artificial Dendritic Spines Wim Pomp, Thomas Schmidt. Leiden University, Leiden, Netherlands. Memory and learning are believed to be regulated by the strength of the connections in the synapses of the 100 billion neurons in the human brain. In the synapse the signal is transmitted between the presynaptic axon and the dendritic spine by neurotransmitters. The number of receptors in the membrane of the dendritic spine defines the strength of a synapse. The persistent increased local concentration of receptors however, is contradicted by the finding of high receptor mobility within the synapse, dependent on spine morphology. The Tuesday, February 10, 2015 observation that synaptic strength correlates with dendritic spine morphology leads to the hypothesis that the mushroom-like shape of dendritic spines functions as a receptor trap. We developed a mimetic system to investigate dendritic spine morphology and its effects on receptor confinement and diffusion. Giant unilamellar vesicles (GUV’s) are made from lipids using electroswelling. To mimic the mushroom-shaped morphologies of dendritic spines, a micromanipulator is used to pull membrane tubes from the GUV lipid bilayer. Trapping capabilities for different spine morphologies are assessed by tracking quantum dots attached to membrane lipids, thus mimicking receptors. Results show a strong dependence of escape times on GUV morphology, as quantified by GUV radius and tube length. Instead of a trivial quadratic dependence of escape times on GUV radius we find a powerlaw dependence with an exponent of 2.85. This confirms the idea that receptors can be trapped by the morphology of a dendritic spine. Therefore the connection strength of a mushroom-shaped dendritic spine is much more stable than the strengths of stubby shaped dendritic spines. Platform: Protein Structure and Conformation III 1724-Plat Transmembrane Signaling through a Bacterial Heme Transporter Halina Wojtowicz1,2, Ada Prochnicka-Chalufour1,2, Idir Malki1,2, Catherine Simenel1,2, Muriel Delepierre1,2, Nadia Izadi-Pruneyre1,2. 1 NMR of Biomolecules Unit, Structural Biology and Chemistry Department, Pasteur Institute, Paris, France, 2CNRS UMR, Paris, France. Bacteria use diverse signaling pathways to control gene expression in response to external stimuli. In Gram-negative bacteria, the binding of some nutrients is sensed by their specific outer membrane transporter. A cascade of molecular interactions between several proteins, located in three subcellular compartments, is then used to send this signal from outside to inside the bacteria and upregulate the expression of genes related to the acquisition of these nutrients. We study a heme acquisition system (Has) developed by several commensal and pathogenic bacteria to acquire heme as iron source. Using multidisciplinary approach (NMR, Xray, SAXS and Electron Microscopy) we have determined the structure of multiprotein complexes involved in the Has signaling pathway. Furthermore, we have recently shown, for the first time, that a partially folded protein is involved in this process 1,2,3,4,5. Our current data represent the first detailed characterization of this type of bacterial signaling. Ref: 1:Krieg S et al 2009 PNAS; 2:Caillet-Saguy S et al JACS 2009; 4: Cardoso de Amorim et al PlosOne 2013; Malki et al PlosOne 2014.Wojtowicz et al, in preparation. 1725-Plat Tom1 Modulates the Endosomal Function of Tollip via a Folding-UponBinding Mechanism Shuyan Xiao1, Mary K. Brannon1, Geoffrey S. Armstrong2, Kristen Fread1, Jeffrey Ellena3, John H. Bushweller3, Carla V. Finkielstein4, Daniel G.S. Capelluto1. 1 Protein Signaling Domains Laboratory, Biological Sciences, Virginia Tech, Blacksburg, VA, USA, 2Chemistry and Biochemistry, University of Colorado, Boulder, CO, USA, 3Chemistry, University of Virginia, Charlottesville, VA, USA, 4Integrated Cellular Responses laboratory, Biological Sciences, Virginia Tech, Blacksburg, VA, USA. Many cellular signaling processes require internalization of ubiquitylated cargo via endocytosis and early endosomes are the first sorting station for vesicular cargo. Adaptor protein complexes modulate signaling output in early endosomes by triggering cargo sorting for lysosomal degradation. Tollip, through its C2 domain, associates with endosomal membranes via phosphatidylinositol 3-phosphate (PtdIns(3)P) and recruits ubiquitylated cargo to these compartments via its C2 and CUE domains. Interestingly, binding of Tollip to PtdIns(3)P is inhibited by ubiquitin. Tom1, through its GAT domain, is recruited to endosomes by binding to cargo and the Tollip TBD region by an unknown mechanism. NMR data revealed that Tollip TBD is a natively unfolded domain that partially folds when bound to Tom1 GAT. Furthermore, the association of Tom1 to Tollip inhibits Tollip’s binding to PtdIns(3)P. We hypothesize that Tom1 GAT binding to Tollip TBD causes conformational changes in Tollip that impairs its binding to PtdIns(3)P, increasing its commitment to protein sorting. Tollip TBD-Tom1 GAT forms a stable heterodimer, whose association is mainly driven by hydrophobic contacts of high affinity. Nuclear spin relaxation studies demonstrate that the N-terminal half structure of Tollip TBD, which contacts Tom1 GAT, is ordered, whereas the C-terminal half is highly unstructured. Ubiquitin binds to Tom1 GAT at a site that does not overlap with that for the Tollip TBD following a fast exchange regime. Cytosolic 343a Tom1 is recruited to endosomal compartments when co-expressed with Tollip in mammalian cells, but mutations, which reduce 90,000-fold the association of these proteins, revert this effect. Accordingly, we propose that association of Tom1 to Tollip facilitate Tollip release from endosomal membranes, allowing Tollip to commit to endosomal cargo trafficking. I respectfully request the Committee consider our work for oral presentation based on its interest to a broad audience. 1726-Plat X-Ray Structure of a Calcium Activated TMEM16 Lipid Scramblase Janine D. Brunner, Novandy K. Lim, Stephan Schenck. Departement of Biochemistry, University of Zurich, Zurich, Switzerland. The TMEM16s or anoctamins constitute a class of eukaryotic membrane proteins that in mammals contain ten members with high sequence conservation. Despite their close relationship these proteins are characterized by a remarkable functional diversity. The family includes the long sought-after Ca2þ-activated chloride channels (TMEM16A and B) but also cation channels and lipid scramblases that support the exchange of lipids between the inner and outer leaflets of the bilayer in an ATP-independent manner. As part of the blood coagulation process, TMEM16F triggers the exposure of phosphatidylserine in blood platelets upon activation by Ca2þ. TMEM16C, D, G and J were suggested to work as scramblases as well, but with variable characteristics. Although we have by now gained considerable insight into the functional properties of certain family members, their architecture and the relation to mechanisms of action were so far unknown. Here we present the crystal structure of nhTMEM16, a fungal family member that operates as a Ca2þactivated lipid scramblase. Each subunit of the homodimeric protein contains ten transmembrane helices and a hydrophilic membrane-traversing cavity that resembles a ‘spiral staircase’ and is exposed to the lipid bilayer as a potential site of catalysis. This cavity harbors a conserved Ca2þ-binding site located within the hydrophobic core of the membrane. Ca2þ binding by six residues, five of which carry a negative charge, controls the activation of scrambling in nhTMEM16. A triple mutant of residues in this site shows only weak scrambling activity that is no longer enhanced by Ca2þ.The structure thus reveals the general architecture of the family and its mode of Ca2þ-activation. It provides insights into potential scrambling mechanisms and will furthermore serve as a framework to unravel the conduction of ions in certain TMEM16 proteins. 1727-Plat Autophagy: Solution Structure of the Atg17-Atg29-Atg31-Atg1-Atg13 Complex Juergen Koefinger1, Michael J. Ragusa2,3, Gerhard Hummer1, James H. Hurley2. 1 Department of Theoretical Biophysics, Max Planck Institute of Biophysics, Frankfurt am Main, Germany, 2Department of Molecular and Cell Biology, California Institute for Quantitative Biosciences, University of California, Berkeley, CA, USA, 3Laboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, USA. Autophagy is a complex bulk clearance mechanism that collects cellular material intended for degradation. In yeast, this highly conserved machinery comprises about 40 autophagy-related (Atg) proteins, most of which form multiprotein complexes. We elucidated the structure of the Atg17-Atg29-Atg31-Atg1Atg13 complex in solution using small-angle X-ray scattering experiments (SAXS), coarse-grained simulations, and the ensemble refinement of SAXS (EROS) method. Our model consists of rigid domains based on the crystal structures of the Atg17-Atg29-Atg31 complex and of the Atg1-Atg13 complex. The modeling and simulation of disordered regions, which have not been resolved in the crystal structures, is crucial for the determination of the solution structures. Specifically, the Atg17 binding site on Atg13 has not been resolved and is located on such a disordered part of the protein. We therefore modeled disordered regions as flexible chains and link the Atg17 proteins and Atg13 proteins accordingly. We performed coarse-grained simulations for various multiprotein complex topologies and refined the resulting ensembles of structures using EROS. The resulting structural models account consistently for the SAXS measurements of the Atg17Atg29-Atg31-Atg1-Atg13 supercomplex and several of its subcomplexes. 1728-Plat Structural Studies of G-Alpha-Q Signaling Veronica G. Taylor1, Elena Kondrashkina2, Paige Bommarito3, George Lund4, Tomasz Cierpicki4, John J.G. Tesmer5. 1 Biophysics, University of Michigan, Ann Arbor, MI, USA, 2Advanced Photon Source, Argonne National Laboratory, Argonne, IL, USA, 3Life Sciences Institute, University of Michigan, Ann Arbor, MI, USA, 4Pathology, University of Michigan, Ann Arbor, MI, USA, 5Pharmacology, University of Michigan, Ann Arbor, MI, USA. The heterotrimeric G protein Gaq has been implicated in a variety of cardiovascular processes, such as blood pressure regulation. Structural studies of 344a Tuesday, February 10, 2015 Gaq signaling can lead to a greater understanding of these cardiovascular processes. G protein-coupled receptors activate Gaq, and this allows Gaq to interact with its effectors, which include guanine nucleotide exchange factors (GEFs). Through formation of a complex with one of these effectors, p63RhoGEF, Gaq has been linked to activation of RhoA, a small molecular weight G protein and key actin cytoskeleton regulator. Although the atomic structure of the Gaq-p63RhoGEF-RhoA complex is known, the mechanism of GEF activation is not clear, in part because the structure of the basal conformation of p63RhoGEF has not yet been determined. We are using nuclear magnetic resonance spectroscopy and small angle x-ray scattering to study the solution structure of the basal, inactive p63RhoGEF catalytic core and to confirm the multi-domain interactions with Gaq that were observed in the original crystal structure. Gaq signaling is also regulated by GTPase activating proteins known as regulator of G protein signaling (RGS) proteins. Whereas RGS2 is the only member of this family known to be selective for the Gaq/11 class of heterotrimeric G proteins, multiple RGS family members are selective against Gaq/11, and the molecular basis for this distinction is unknown. We have determined the crystal structure of Gaq in complex with RGS8, which is selective for multiple classes of Ga subunits. This structure adds new insights into the molecular basis of RGS protein selectivity for distinct classes of Ga subunits. 1729-Plat Kinetics and Thermodynamics of Apicomplexa AMA1-RON2Sp Interaction Roberto F. Delgadillo1, Maryse Lebrun2, Martin Boulanger3, Dominique Douguet1. 1 Institut de Pharmacologie Mole´culaire et Cellulaire, Sophia Antipolis, France, 2UMR 5235 CNRS, Universite´ de Montpellier 1 et 2, Montpellier, France, 3Biochemistry & Microbiology, University of Victoria, Victoria, BC, Canada. Plasmodium falciparum and Toxoplama gondii are obligate intracellular protozoan parasites that invade and replicate within host cells. They both require the formation of a tight interaction with the host cell, called Moving Junctions (MJ), for successful infection. It has been shown that the MJ contains two key parasite components: the surface protein Apical Membrane Antigen 1 (AMA1) and its receptor, the Rhoptry Neck Protein (RON) complex, the latter one being targeted to the host cell membrane during invasion. Crystal structures of AMA1 proteins have shown a versatile loop, called domain II loop that extends into domain I likely to hide the RON2 binding site from host immunity. In the present work, we have studied association, dissociation reactions and binding equilibria of PfAMA1 and TgAMA1 reacting with their respective RON2 short peptide ligand. Equally, we have studied a deltaDIIloop-PfAMA1 construct to elucidate the role of this loop upon RON2 peptide binding. The reactions were tracked by fluorescence anisotropy as a function of temperature and concentration and globally fitted to acquire the rate constants to calculate the thermodynamic profile and propose a reaction mechanism. Our results showed that PfAMA1 and TgAMA1 bind to their respective RON2 peptide with the formation of one intermediate in a sequential reversible reaction: A4B4C. The reactions are both enthalpically and entropically favorable upon ligand binding thanks of the DII-loop induced fit folding down over the bound ligand forming a most stable final complex. The half life time of the complex at 25 C is 326s and 1077s for Pf and Tg complexes, respectively. By in vitro-in vivo extrapolation at 37 C, it is compatible with the time frame of erythrocyte invasion by Plasmodium falciparum merozoites. The elucidation of the binding mechanism brings new strategies for ligand discovery against these pharmacologically important targets. 1730-Plat Conformation of the Troponin I C-Terminal Domain in Silico and in vitro: A Consideration of Dynamics in Comparing Simulation and Experiment Lauren Ann Metskas, Elizabeth Rhoades. Yale University, New Haven, CT, USA. The troponin complex acts as a molecular switch in striated muscle cells to regulate myosin attachment to and isomerization on actin filaments in response to changes in calcium concentration. Transitions between the inactive and active states of the thin filament require extensive domain movements and binding exchanges involving the C-terminal domain of cardiac troponin I (TnI-C), believed to be intrinsically disordered in the high-Calcium state [1]. Mutations in TnI-C are associated with hypertrophic cardiomyopathy, highlighting the importance of this domain in regulating cardiac contraction; however, the conformational flexibility of the domain has delayed its characterization compared with the rest of the troponin complex. Here, we use single molecule Fo¨rster resonance energy transfer (smFRET) to probe the global conformation of TnI-C in the high-Calcium state, capitalizing on the technique’s ability to analyze heterogeneous populations. We compare six pairwise distances within TnI-C to outputs from molecular dynamics simulations to gain insight into conformational sampling at finer detail and a faster timescale than experimental measurements allow. We find that simulations are in good agreement with smFRET measurements, but only after simulations are averaged over time. Simulations rarely, if ever, sample an ‘‘ideal’’ conformation matching all experimental measurements simultaneously; this finding highlights the importance of considering timescale when combining simulations and experimental measurements. Using our combined in silico and in vitro approach, we can predict areas of helical propensity and cluster potential global domain conformations in the unbound state, potentially providing mechanistic insight into the coupled binding and folding of this region during muscle relaxation. 1. Julien, O., Mercier, P., Allen, C.N., et al. (2011) Is there nascent structure in the intrinsically disordered region of troponin I? Proteins 79: 1240-1250. 1731-Plat NMR Structural Studies of a 52 kDa Heterocylization Domain of the Yersiniabactin Non-Ribosomal Peptide Synthetase Subrata H. Mishra, Bradley J. Harden, Scott R. Nichols, Dominique P. Frueh. Biophysics and Biophysical Chemistry, Johns Hopkins University, School of Medicine, Baltimore, MD, USA. Nonribosomal peptide synthetases (NRPSs) are modular multi-domain enzymatic systems in bacteria and fungi that synthesize a diverse array of secondary metabolites called nonribosomal peptides (NRPs). NRPs encompass broad biological activity from etiological agents in microbial infections to various pharmaceutical applications. Despite their diversity NRPs are synthesized in a similar iterative manner, where each module of the synthetase adds a single substrate to the growing NRP chain. Chain elongation proceeds via peptide bond formation, catalyzed by condensation domains (C), between substrates tethered onto thiolation domains (T) in sequential modules. Condensation domains are sometimes replaced by cyclization domains (Cy) that carry out both condensation and heterocyclization (e.g. cysteines to thiazolines). NRP chain elongation affected by C (or Cy) and cognate T domain interactions is poorly understood due to lack of molecular details. Multidomain X-ray structures revealed only non-functional inter-domain orientations. Solution NMR studies have highlighted the presence of multiple conformers of excised domains in equilibria suggesting that transient domain interactions driven by conformational selection propel NRP chain elongation. Here, we investigate the molecular interactions between a Cy domain and its two cognate T domains from the NRPS Yersiniabactin synthetase using solution NMR techniques. We have determined the NMR solution structure of this excised 52 kDa Cy domain. New NMR methods were developed to overcome challenges in chemical shift assignments and improving the accuracy of distance constraints in the structure determination of this large protein. The interaction interfaces between the Cy and the T domains were mapped by chemical shift perturbation data from titrations of each T domain individually and sequentially into Cy. Our studies lay the foundation for building a model of this ternary complex to better understand domain interactions at a molecular level in NRP synthesis. Platform: Bioengineering and Biomaterials 1732-Plat Nanoparticle-Induced Membrane Pore Formation Studied with Lipid Bilayer Arrays Jacob Schmidt. Bioengineering, University of California Los Angeles, Los Angeles, CA, USA. Nanoparticles are present environmentally as byproducts of industrial processes and in a wide range of consumer goods. Although toxicity studies of nanoparticles and other nanomaterials have only begun relatively recently, a diverse range of nanoparticles has already been shown to be toxic. Nanoparticle cytotoxicity assays provide little information about the mechanisms of nanoparticle toxicity, which are potentially complex and are not likely the same for diverse nanoparticle species. For this reason, artificial lipid bilayer platforms have begun to be used as model systems for controlled studies allowing variation of experimental parameters not possible with cellular studies, such as membrane and solution composition. Unfortunately, in some cases the low throughput characteristic of lipid bilayer experimentation can limit the experimental scope. We have recently described a lipid bilayer array platform with simultaneous bilayer formation and measurement over a 32-element array with ~80% yield and no operator input following fluid addition.[1] The platform is modular and allows rapid cycling of the apparatus for Tuesday, February 10, 2015 repeated measurement. We have used this platform to measure the interactions of aminated and carboxylated polystyrene nanoparticles and a range metal oxide nanoparticle species with lipid bilayers in a wide variety of experimental conditions, including nanoparticle concentration, bilayer composition, bilayer charge, presence of serum protein, solution ionic strength, and pH. The array format permitted several thousand bilayers to be measured in total with sufficient redundancy to give statistical significance to measured results. Detailed analysis of the electrical measurements shows pore formation that is dependent on electric field, ionic strength, and nanoparticle species. 1. ‘‘Lipid bilayer arrays cyclically formed and measured,’’ Bin Lu, Gayane Kocharyan and Jacob J. Schmidt, Biotechnology Journal 9, 446-451 (2014) 1733-Plat Sticky Patches on Lipid Nanoparticles Generate Binding Geometries that Enable Effective Targeting of Otherwise Untargetable Cancers Michelle Sempkowski1, Yannis Kevrekidis2, Stavroula Sofou1. 1 Rutgers University, Piscataway, NJ, USA, 2Princeton University, Princeton, NJ, USA. The majority of breast cancer patients (70%) have tumors designated as ‘HER2-negative’ (<1þ HER2-expression evaluated by immunohistochemistry or < 200,000 HER2-copies per cell). For these patients there are no targeted therapeutic options utilizing the HER2 receptor. The ability of conventionally targeted nanoparticles for specific targeting stops to hold on cancer cells expressing less than 200,000 copies of HER2 per cell or less than two receptors per nanoparticle’s projected area (for particles of 100 nm in-diameter). This geometry corresponds to the limit of multivalent interactions (avidity) loosely defined as multiple contacts between neighboring same-cell receptors with ligands from a single nanoparticle. An alternative therapeutic approach is needed, therefore, to enable selective targeting and effective killing of cancer cells with low or too low HER2 expression. Towards this goal we designed targeted lipid nanoparticles (vesicles) that contain HER2-targeting short peptides densely conjugated (for high local multivalency) within sticky patches. Sticky patches are phase-separated raft-like lipid-domains of high local multivalency which is induced by preferential partitioning of peptide-functionalized lipids. To enable selectivity in binding, sticky patches are exclusively triggered to form in mildly acidic environments matching the tumor interstitium. Lipid phase-separation with lowering pH is a result of the interplay of decreasing (pH-tunable) electrostatic repulsion and attractive hydrogen bonding among the domainforming lipids. We show that lipid nanoparticles with sticky patches selectively associate with and kill HER2-negative and triple negative breast cancer cells (MCF-7 and MDA-MB-231, respectively, with 60,000 and 90,000 HER2-copies per cell) while do not affect cardiomyocytes and breast normal cells. Systematic studies of association, dissociation and internalization rates of nanoparticles by cells will be presented, and a mechanistic mathematical model will be discussed with the aim to explain the observed high avidity. 1734-Plat Controlled Activation of Protein Rotational Dynamics using Smart Hydrogel Tethering Yijia Xiong1, Brenda M. Beech2, Curt B. Boschek2, Cheryl L. Baird2, Diana J. Bigelow2, Kathleen McAteer3, Thomas C. Squier1. 1 Basic Medical Sciences, Western University of Health Sciences, Lebanon, OR, USA, 2Biological Sciences, Pacific Northwest National Laboratory, Richland, WA, USA, 3Biological Sciences, Washington State Universtiy Tri-Cities, Richland, WA, USA. Stimulus-responsive hydrogel materials that stabilize and control protein dynamics have the potential to enable a range of applications that take advantage of the inherent specificity and catalytic efficiencies of proteins. Here we describe the modular construction of a hydrogel using an engineered calmodulin (CaM) within a polyethylene glycol (PEG) matrix that involves the reversible tethering of proteins through an engineered CaM-binding sequence. For these measurements, maltose binding protein (MBP) was isotopically labeled with [13C] and [15N], permitting dynamic structural measurements using TROSY-HSQC NMR spectroscopy. Protein dynamics are suppressed upon initial formation of hydrogels, with concomitant increases in protein stability. Relaxation of the hydrogel matrix following transient heating results in enhanced protein dynamics and resolution of substrate-induced large-amplitude domain rearrangements. Our results demonstrate an ability to take advantage of the conformational sensitivities of hydrogel materials to activate protein dynamics upon transient temperature increases. Such an approach permits storage of proteins in an immobilized state prior to their activation, and contributes to important applica- 345a tions that can take advantage of the specificity of proteins for a range of sensing and chemical transformation applications. For example, single chain antibodies are shown to be dramatically stabilized against denaturation by urea, enabling their long term use for sensing applications. These smart materials possess optimized mass transfer properties (due to their high water content) and provide important avenues to detect ligands so as to link binding to material responses (e.g., proteolysis or other types of chemical transformation using the catalytic specificities of enzymes). Supported by DTRA under HDTRA1-08-10-BRCWMD award 10027-2828. NMR measurements at the Environmental Molecular Sciences Laboratory, a national scientific user facility supported by DOE’s Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory (PNNL). 1735-Plat Immobilization of Proteins on Chemically Modified Germanium Investigated by ATR-FTIR Jonas Schartner, Konstantin Gavriljuk, Andreas Nabers, Klaus Gerwert, Carsten Ko¨tting. Biophysics, Biology & Biotechnology, Bochum, Germany. The attenuated total reflection fourier transform infrared spectroscopy (ATRFTIR) allows a detailed analysis of surface attached molecules, including their secondary structure, reaction mechanism, orientation and interaction with small molecules or proteins.1 The aim of our study is the development of a universal immobilization technique on germanium for all kinds of proteins. We recently showed the specific immobilization of N-Ras and Photosystem I on a silane modified germanium surface.1 We now present a new approach employing thiol chemistry on germanium.2,3 On one hand germanium crystals provide a great signal-to-noise ratio in ATR-FTIR. On the other hand protein immobilization via thiol chemistry is well-established because it is standard for modifications of gold surfaces e.g. in surface plasmon resonance. Here we combine the best of both worlds and report on germanium surface functionalization with different thiols which allowed for specific immobilization of histidine-tagged proteins with over 99% specific binding. The great advantage of using thiols in comparison with silanes is that a huge variety of thiols with functional groups for many kinds of protein immobilizations are readily available and the higher stability. Nativity of protein folding was confirmed by secondary structure analysis. Stimulus induced difference spectra were obtained for immobilized Channelrhodopsin 2, the small GTPase N-Ras and the phosphocholine-transferase AnkX, which demonstrated protein function at the atomic level.4 Protein activity was observed for Channelrhodopsin 2 for over several days.4 1: Schartner J. et al., J. Am. Chem. Soc., 2013, 135, 4079-4087 2: Hanrath, T. & Korgel, B. A., J. Am. Chem. Soc., 2004, 126, 15466-15472 3: Han, S. et al. J. Am. Chem. Soc., 2001, 123, 2422-2425 4: Schartner J., et al., ChemBioChem, 2014, accepted 1736-Plat Use of Short Amyloidogenic Peptides in Protein-Ligand Detection Systems Gabriela M. Guerra, So´nia Gonc¸alves, Nuno C. Santos, Ivo C. Martins. Biomembranes Unit, Institute of Molecular Medicine (IMM), Lisbon, Portugal. Amyloid fibers, often associated with many human degenerative diseases (such as Alzheimer’s and Parkinson’s disease), may also have physiological roles, having even been suggested as potential novel biomaterials [1-2]. Since it is now clear that the amyloid fibers are much less toxic than their precursor aggregates [3], the interest for amyloidogenic species in nanosensing and proteinligand detection sytsems increased dramatically [2]. Amyloid fibers in general share a common b-sheet rich architecture that is behind their exceptional stability, mechanical strength and resistance to degradation, which in nanotechnology makes them excellent nanomaterials candidates [1]. The potential to form amyloids (and other protein/peptide aggregates) can be predicted from the peptide amino acids sequence [1, 2]. Here, we used different amyloid peptide sequences to evaluate, by AFM, circular dichroism and FTIR spectroscopic approaches under different conditions, which type of amyloid species would be formed (namely amyloid oligomers, protofibrils or fibrils) at different times of incubation (24 hours, 72 hours and 2 weeks). AFM, CD and FTIR data taken together indicate that amyloid-based nanotechnology approaches may be successfully employed. References (* stands for the presenting author own work) [1] Cherny & Gazit, 2008, Angew Chem Int Ed Engl, 47:4062 [2] Hauser et al., 2014. Chem Soc Rev, 43:5326* [3] Martins et al., 2008. EMBO J, 27:224* [4] Maurer-Stroh et al., 2010, Nat Methods, 7:237* 346a Tuesday, February 10, 2015 1737-Plat Ice Growth Control with Ice-Binding Proteins Ido Braslavsky1,2, Ran Drori1, Yeliz Celik2, Peter L. Davies3. 1 Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot, Israel, 2Department of Physics and Astronomy, Ohio University, Athens, OH, USA, 3Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON, Canada. Ice-binding proteins (IBPs) depress the freezing point of body fluids below the melting point, resulting in a thermal hysteresis (TH) that prevents freezing of the organism. The potential of these proteins in the medical sector, in cryopreservation, in the frozen food industry, and in agriculture is enormous. We are investigating the mechanism by which IBPs interact with ice surfaces and inhibit ice growth and recrystallization. We have developed novel methods for these studies, including fluorescence microscopy techniques combined with temperature-controlled microfluidic devices. These techniques have enabled the replacement of the IBP solution surrounding an IBP-bound ice crystal by buffer, without losing the bound IBP or the TH activity. Our results show the irreversibility of the protein:ice interactions and the indirect dependence of TH activity on the protein concentration in solution. We found that the dynamics of the interactions with ice vary dramatically between different types of IBPs. From our results and other recent developments a new understanding of the mechanisms by which various IBPs act is emerging. This understanding is critical for the successful use of IBPs in cryobiological applications. Supported by the European-Research-Council (ERC), the National-ScienceFoundation (NSF), and the Israel-Science-Foundation (ISF), Canadian Institutes of Helth (CIHR), The Lady Davis Foundation, and the Canada Research Chair program. Website: http://www.agri.huji.ac.il/~braslavs/ References Drori et al, Ice-Binding Proteins that Accumulate on Different Ice Crystal Planes Produces Distinct Thermal Hysteresis Dynamics, J. R. Soc. Interface (2014), 11:20140526. Celik, et al, Microfluidic experiments reveal that antifreeze proteins bound to ice crystals suffice to prevent their growth, PNAS (2013) 110, 1309-1314. 1738-Plat A Two-Color Non-Muscle SERCA FRET Sensor for Diabetes Drug Discovery Using Fluorescence Lifetime Detection Tory Schaaf1, Ji Li1, Rocio Foncea1, Simon Gruber1, Kurt Peterson2, Karl Petersen1, Cornea Razvan1, Greg Gillispie1,2, David Bernlohr1, David Thomas1. 1 Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN, USA, 2Fluorescence Innovations, Inc., Minneapolis, MN, USA. We have developed intramolecular FRET sensors capable of detecting cytoplasmic headpiece movements of human SERCA (sarco/endo-plasmic reticulum calcium ATPase) in live-cell assays, including a non-muscle SERCA2b isoform to be used in drug discovery for treatment of diabetes. Two fluorescent proteins, clover (green) and mRuby2 (red) were directly fused to selected locations on human SERCA1a (skeletal muscle), 2a (cardiac muscle), and 2b (non muscle), based on a previously reported SERCA2a construct (Gruber et al., J. Biol. Screening, 2014), and expressed stably in HEK cells. We have used these cells in a novel fluorescence lifetime plate reader (FLT-PR) to screen small-molecule libraries, to discover modulators of SERCA structure and function. The present study focuses on SERCA2b, with the goal of obtaining small molecules that activate SERCA in non-muscle cells. Since recent reports indicate that SERCA overexpression in non-muscle cells can alleviate Type II diabetes, we seek small-molecule SERCA activators for the same purpose. The small-molecule modulators identified in the high-throughput FRET screen were examined for their ability to affect SERCA’s function, through assays of ATPase and calcium pumping activities. In order to obtain functional data more directly related to Type II diabetes, we tested the compound’s alleviation of endoplasmic reticulum stress in 3T3-L1 adipocytes, using an XF24 Extracellular Flux Analyzer to measure mitochondrial function after inducing ER stress with the inflammatory cytokine TNF-a. While this study is designed to find activators of SERCA2b for treatment of diabetes, constructs based on other SERCA isoforms show promise in targeted therapeutics for muscular dystrophy (SERCA1a) and heart failure (SERCA2a). 1739-Plat A Novel Molecular Ruler between Fluorescent Proteins Gary C.H. Mo, Jin Zhang. Pharmacology, Johns Hopkins University School of Medicine, Baltimore, MD, USA. Fo¨rster Resonance Energy Transfer (FRET) is a phenomenon that allows the direct measurement of molecular events through macroscopic observations. The simplicity of FRET translates to its robust performance in complex environments such as living cells, making it immensely powerful in molecular biology. Here, we demonstrate an analogous molecular event between fluorescent proteins, which relies on a different mechanism but shares many parallels with FRET. We demonstrate an exquisite distance sensitivity that allows us to distinguish the lengthening of a protein linker by 1 helical turn in live cells. Further, coupling this discovery with known molecular switches forms the basis of a novel class of biosensors. We therefore report the superresolution imaging of kinase activity and protein-protein interaction for the first time. We utilize one such biosensor to scrutinize the spatial activity architecture of cAMPdependent protein kinase (PKA). Our results directly confirm the compartmentalization of PKA signaling, and illuminate their characteristics in adherent and migrating cells. In summary, we report a novel molecular ruler that parallels FRET in many aspects and will be useful in a similarly wide range of applications. Platform: Protein Fold Stability 1740-Plat The Folding of SasG: A Long and Remarkably Strong Monomeric Protein Responsible for Biofilm Formation is a Highly Cooperative System Dominika T. Gruszka1, Fiona Whelan2, Emanuele Paci3, David J. Brockwell3, Jennifer R. Potts2, Jane Clarke1. 1 Department of Chemistry, University of Cambridge, Cambridge, United Kingdom, 2Department of Biology, University of York, York, United Kingdom, 3School of Molecular and Cellular Biology, University of Leeds, Leeds, United Kingdom. SasG has a long repeat region made up of identical repeating E and G5 domains. Although the domains themselves are relatively unstable (indeed E domains on their own are unfolded), the cooperative folding of the domains results in formation of molecules that are long and remarkably mechanically resistant. We have used small angle X-ray scattering and mechanical unfolding methods, combined with simulations, to show that SasG constructs of physiological length are indeed monomeric, highly extended and mechanically strong. Obligate folding cooperativity of the intrinsically disordered E domain couples spatially separate G5 domains both thermodynamically and structurally, creating a superstructure that supersedes the domain architecture. Our findings provide a simple solution for the efficient assembly of mechano-resistant elongated structures of tunable length from a single polypeptide chain and have significant potential for the development of novel biomaterials. 1741-Plat Putting on the Squeeze: Solution NMR Investigations of Protein Structure and Hydration under High Pressure Nathaniel V. Nucci1, Brian Fuglestad2, Connie Liao2, Evangelia A. Athanasoula2, A. Joshua Wand2. 1 Physics & Astronomy and Biomedical & Translational Sciences, Rowan University, Glassboro, NJ, USA, 2Biochemistry and Biophysics, University of Pennsylvania, Philadelphia, PA, USA. It is well known that high hydrostatic pressures can induce the unfolding of proteins. The physical underpinnings of this phenomenon have been investigated extensively but remain controversial. Changes in solvation energetics due to applied hydrostatic pressure have been a commonly proposed mechanism for unfolding, but recent studies have provided strong evidence that elimination of void volumes in the native folded state is a principal determinant. Here we use the cavity-containing L99A mutant of T4 lysozyme to examine the pressure unfolding of a multi-domain protein using solution NMR. The cavity-containing C-terminal domain completely unfolds at moderate pressures while the N-terminal domain remains largely structured to high pressures. This pressure response is completely suppressed by benzene binding to the hydrophobic cavity. These results contrast to the pseudo wild type protein, which has a residual cavity volume very similar to that of the L99A-benzene complex but shows extensive subglobal reorganizations with pressure. Encapsulation of the L99A mutant in the aqueous nanoscale core of a reverse micelle suppresses the pressure-induced unfolding transition due to the volume restriction and promotes high-pressure filling of the cavity with water. This result indicates that hydration of the hydrophobic cavity is more energetically unfavorable than global unfolding. Overall these observations point to a range of cooperativity and energetics in the pressure response of proteins and illuminate the fact Tuesday, February 10, 2015 that small changes in physical parameters can significantly alter this response. Supported by NSF grant MCB- 115803 and by NIH postdoctoral fellowship GM087099 to N.V.N. 1742-Plat Effects of Crowding, Osmolytes, Temperature and Pressure on the Interaction Potential of Dense Protein Solutions Roland Winter. TU Dortmund University, Dortmund, Germany. We studied the effect of pressure on the structure and intermolecular interactions of dense lysozyme solutions in various cosolvent mixtures and upon addition of various Hofmeister anions using small-angle X-ray scattering in combination with liquid-state theoretical approaches [1-3]. Supplementary thermodynamic information was obtained by employing calorimetric techniques, densitometry and ultrasound velocimetry. We show that the particular structural properties of water and specific ion effects play a crucial major role in protein stabilisation, notably under high hydrostatic pressure conditions. Also the effect of confinement on the solvational properties and intermolecular interaction of proteins was studied, including the effects of self-crowding and macromolecular crowders on the temperature-pressure stability diagram of proteins [4]. We also discuss the effect of pressure on the second virial coefficient and how pressure can be used to control and fine-tune protein crystallization. Moreover, we present results on the phase behavior of dense lysozyme solutions in the liquid-liquid phase separation region. A re-entrant liquid-liquid phase separation region has been discovered at elevated pressures, which originates in the pressure dependence of the solvent-mediated protein-protein interactions [3]. [1] M. A. Schroer, J. Markgraf, D. C. F. Wieland, C. J. Sahle, J. Mo¨ller, M. Paulus, M. Tolan, R. Winter, Phys. Rev. Lett. 106 (2011) 178102 [2] M. A. Schroer, Y. Zhai, D. C. F. Wieland, C. J. Sahle, J. Nase, M. Paulus, M. Tolan, R. Winter, Angew. Chem. Int. Ed. 50 (2011) 11413 [3] J. Mo¨ller, S. Grobelny, J. Schulze, S. Bieder, A. Steffen, M. Erlkamp, M. Paulus, M. Tolan, R. Winter, Phys. Rev. Lett. 112 (2014) 028101 [4] M. Erlkamp, S. Grobelny, R. Winter, Phys. Chem. Chem. Phys. 16 (2014) 5965 1743-Plat A Multiscale Model for pH-Dependent Folding and Binding of a Conditionally Disordered Chaperone Logan S. Ahlstrom, Sean M. Law, Alex Dickson, Charles L. Brooks III. Department of Chemistry, University of Michigan, Ann Arbor, MI, USA. The bacterial acid stress-sensing chaperone HdeA loses structure to gain function. As enteropathogenic E. coli pass through the severely acidic environment of the mammalian stomach, HdeA transitions from an inactive, folded dimer to chaperone-active, unfolded monomers to protect against the acidinduced aggregation of periplasmic proteins. Toward achieving an atomiclevel mechanistic understanding of the acid stress response of HdeA, we develop a multiscale modeling approach to capture its pH-dependent thermodynamics. Our approach utilizes pKa calculations from all-atom constant pH molecular dynamics simulations to alter the coarse-grained model for representing different pH environments. Changes in the thermodynamics of binding as a function of pH are explored using the efficient ‘‘Hamiltonian mapping’’ reweighting formalism. We propose new features of the pHsensing mechanism of HdeA that can be directly tested by experiment. Namely, our model predicts that HdeA is maximally stable under mildly acidic conditions and that a partially unfolded dimeric intermediate may contribute to substrate binding. Our multiscale approach is general such that it can be applied toward understanding pH-dependent functional transitions in other systems and sets a foundation from which to construct models of HdeA-substrate interaction. 1744-Plat Structural Origin of Landscape Roughness in Protein Folding from SingleMolecule FRET and All-Atom Molecular Dynamics Simulations Hoi Sung Chung1, Stefano Piana-Agostinetti2, David E. Shaw2, William A. Eaton1. 1 Laboratory of Chemical Physics, NIDDK/NIH, Bethesda, MD, USA, 2D. E. Shaw Research, New York, NY, USA. Folding of most single-domain proteins has been successfully described by diffusion on a one-dimensional (1D) free energy surface. Although the 1D surface is smooth, there are many local minima in the underlying energy landscape, giving rise to landscape ‘‘roughness’’. According to Kramers’ reaction-rate theory, roughness slows folding kinetics by reducing the diffusion coefficient at the top of the free energy barrier that separates folded and unfolded states. By measuring the transition-path time (tTP) from a maximum 347a likelihood analysis of photon trajectories in single molecule FRET experiments, we have recently shown that the Kramers diffusion coefficient for a designed a-helical protein, a3D, is markedly reduced (Chung and Eaton, Nature,2013). To discover the structural origin of this slow diffusion, we have combined additional single-molecule FRET measurements with allatom molecular dynamics (MD) calculations. a3D contains 12 negativelycharged and 10 positively-charged side-chains. Analysis of the transition paths in the MD simulations shows that many non-native salt-bridges form during the folding transition path, suggesting them as the structural origin of long tTP . To test this idea, we lowered the pH to neutralize the carboxylates and eliminate salt-bridges, which increased the folding rate by about 10-fold and significantly reduced tTP. Although it was only possible to determine an upper bound for tTP, even at the highest possible solvent viscosity (15 cP), simulations of photon trajectories suggested that most, if not all, of the increase in folding rate could be accounted for by a decreased tTP and an increased Kramers diffusion coefficient. Neutralizing the carboxylates in MD simulations also increases the folding rate and diffusion coefficient and decreases tTP. These results provide the first quantitative glimpse of the effect of specific intra-molecular interactions on barrier crossing dynamics in protein folding. 1745-Plat Resolving Cooperative Interactions in Protein Folding Jacob D. Marold1, Thuy P. Dao2, Tural Aksel3, Doug Barrick1. 1 Biophysics, Johns Hopkins University, Baltimore, MD, USA, 2Chemistry, Syracuse University, Syracuse, NY, USA, 3Biochemistry, Stanford University, Stanford, CA, USA. Understanding the intricate relationships between protein sequence, structure, and stability remains a challenge. One major obstacle in understanding these relationships is the identification of interactions that couple elements of structure; these interactions are not readily apparent from structure. Recently, the application of nearest-neighbor models to repeat protein folding have begun to provide insights into how these systems distribute their energy within, and between units of protein structure1,2,3. Consensus ankyrin and 34-residue cTPR protein (aka c34PR) folding can be described using two terms: the intrinsic energy of individual repeats (DGi), and the interfacial energy between adjacent repeats (DGi,iþ1). Here, we extend this approach in two ways using two experimental TPR-like systems. First, we dissect the whole-repeat nearest-neighbor model by representing (and resolving) the subunits of the nearest-neighbor model into each of the two helices (‘‘A’’ and ‘‘B’’) of each repeat. This extended Ising analysis allows us to quantify coupling energies within and between repeats, as well as local stability differences of the A and B repeat helices. Second, we explore a new, longer class of TPR-like proteins with 42 residues per repeat (42PRs, as opposed to 34 residues for the founding TPR sequence motif), to determine how variation in helix length and interface structure affects intrinsic and coupling energy. By varying the length of constructs in half-repeat increments, we find energetic heterogeneity at both the intrinsic and interfacial level. Although stabilities are more homogeneously distributed in the c34PR series (DGAB and DGBA as well as DGA and DGB are nearly isoenergetic), the distribution is more heterogeneous within 42PRs, with a greater energetic separation between intrinsic and interfacial energies. This leads to greater folding cooperativity within the 42PR series. [1] Wetzel and Pluckthun, JMB [2] Aksel, Structure [3] Kajandar et al, JACS 1746-Plat Mapping the Mechanism of Fast Protein Folding with Multiple Probes Taras V. Pogorelov1, Maxim B. Prigozhin2, Shu-Han Chao3, Martin Gruebele4. 1 School of Chemical Sciences, Department of Chemistry, Beckman Institute, University of Illinois, Urbana, IL, USA, 2Department of Chemistry, University of Illinois, Urbana, IL, USA, 3Department of Physics, University of Illinois, Urbana, IL, USA, 4School of Chemical Sciences, Chemistry, Physics, Biophysics, Beckman Institute, University of Illinois, Urbana, IL, USA. Unlike reactions of small organic molecules, it is not always possible to accurately describe the process of protein folding by a single reaction coordinate. We enhance the structural resolution of microsecond protein folding experiments by introducing new fluorescent inter-helical contact probes into the model protein lambda repressor fragment 6-85. We design two new mutants containing tryptophan and tyrosine residues that interact in the native state. Temperature jump relaxation experiments on these new mutants, in 348a Tuesday, February 10, 2015 combination with a previously available variant, show folding kinetics that is in correspondence with the results obtained from autocorrelation analysis of single-trajectory full-atom molecular dynamics simulations for a similar mutant. Closer investigation of existing all-atom computational data suggests that helix 2 of lambda 6-85 is involved in a short-lived off-pathway trap, which is in agreement with experimental data. Our work demonstrates that a match between fast protein folding experiments and molecular dynamics simulations can be extended to several reaction coordinates to obtain experimental confirmation of deviations from two-state folding behavior even for very simple folding reactions. As computation becomes more affordable, it will be possible to simulate both the new probes and any mechanistic deviations that insertion of the probe causes in experiment. 1747-Plat Photobleaching and Stability of Red Fluorescent Proteins Mengyang Xu1, Deepu K. George1, Ralph Jimenez2, Andrea G. Markelz1. 1 Department of Physics, University at Buffalo, SUNY, Buffalo, NY, USA, 2 Department of Chemistry and Biochemistry, University of Colorado, Boulder, CO, USA. Fluorescent proteins (FPs) are used routinely to visualize structural organization and protein dynamics. Compared with other mutants, there is a large demand for red FPs (RFPs) due to the higher transparency of cells and tissue at longer wavelengths. However, the application of RFPs is limited by the increased susceptibility to photobleaching [1]. A possible mechanism for this may be a decrease in structural stability of the beta-barrel, leading to dark state conversion of the chromophore or oxygen access. To characterize the relation between photostability and structural stability, we use temperaturedependent terahertz (THz) time-domain spectroscopy [2]. Temperature dependent terahertz absorbance measurements were made between 80-270K as a function of photobleaching for mCherry, TagRFP-T and mOrange2 RFP samples. We find that: 1) the absolute THz response follows the thermal stability, as defined by the melting temperature; 2) the protein dynamical transition temperature [3] also follows the thermal stability; 3) the thermal stability increases with photobleaching and 4) the photostability does not follow the thermal stability. The THz sensitivity to thermal stability is verified by CD measurements. The higher stability with photobleaching is surprising, but could possibly be a driving force toward the photobleached state. Our result provides additional insight into photobleaching mechanism, and introduces a way to estimate the qualities of FPs. Platform: Voltage-gated K Channels II 1748-Plat Emerging Role for KCNQ1 in Ischemia-Induced Neuronal Death Kelly A. Aromolaran1, Jee-Yeon Hwang1, Thomas V. McDonald2, R. Suzanne Zukin1. 1 Department of Neuroscience, Albert Einstein College of Medicine, Bronx, NY, USA, 2Departments of Medicine and Molecular Pharmacology, Albert Einstein College of Medicine, Bronx, NY, USA. The slowly activating component of the delayed rectifier potassium current, IKs, composed of KCNQ1-KCNE1 channel subunits, is an important contributor to normal cardiac repolarization. Emerging evidence indicates that KCNQ1 subunits are also expressed in brain, but its precise physiological role in neuronal function has not been investigated. Potassium channels are key players in ischemia-induced death of hippocampal CA1 pyramidal neurons which, in turn, leads to impaired cognition. REST, a gene silencing transcription factor, is up-regulated in global ischemia and contributes to neuronal death. Recently we showed that REST is enriched at the promoter of the KCNQ1 channel and that KCNQ1 mRNA and protein expression are increased in post-ischemic CA1 neurons. Additionally, functional KCNQ1 channels were identified in cultured neurons using KCNQ1-specific inhibitors Chromanol 293B and JNJ-303 and induction of oxygen-glucose deprivation (OGD) induced a marked increase in KCNQ1 currents (~34%, p > 0.02), evident at 48 h post-ischemia. To assess a possible role for KCNQ1 in ischemiainduced neuronal death, we induced OGD in dissociated hippocampal neurons in the absence and presence of Chromanol 293B (100 mM) and treated with propidium iodine. Inhibition of KCNQ1 markedly diminished cell death, assessed at 24 h (21%, p > 0.002) and 48 h (28%, p > 0.001) suggesting that an upregulation of KCNQ1 channels may contribute to ischemia-induced neuronal death. To examine a possible role for REST in ischemia-induced up-regulation of KCNQ1, we performed single-locus ChIP which revealed enrichment for REST by ~2-fold and a decrease in trimethylation of lysine 27 on histone 3 (H3K27me3), an epigenetic mark of gene repression at the KCNQ1 promoter, consistent with the increase in KCNQ1 expression after ischemia. Our findings reveal, for the first time, a role for REST in KCNQ1 expression in response to ischemic insults. 1749-Plat Spectroscopic and Biochemical Studies of TRIP8b Regulation of HCN Channels John R. Bankston1, Hannah A. DeBerg1, Joel C. Rosenbaum2, Peter S. Brzovic2, Stefan Stoll3, William N. Zagotta1. 1 Physiology and Biophysics, University of Washington, Seattle, WA, USA, 2 Biochemistry, University of Washington, Seattle, WA, USA, 3Chemistry, University of Washington, Seattle, WA, USA. TRIP8b, an accessory subunit of hyperpolarization-activated cyclic nucleotidegated (HCN) channels, alters both cell surface expression and cyclic nucleotide dependence of these channels. The mechanism through which TRIP8b exerts these dual effects is still poorly understood. Besides binding the terminal three residues of HCN channels, TRIP8b also binds directly to the cyclic nucleotide-binding domain (CNBD). A small central portion of TRIP8b, termed TRIP8bcore, is involved in this interaction. Binding of TRIP8bcore to the CNBD dramatically reduces the effects of cAMP on the channel. Using spectroscopic and biochemical techniques, we sought to understand how and where TRIP8b binds to the CNBD and how it reduces the cyclic nucleotide dependence of HCN channels. To closely examine the binding of TRIP8bcore to the CNBD, we used double electron-electron resonance (DEER) at Q-band frequencies to study conformational changes in the soluble CNBD of HCN2 in the presence of TRIP8b and cAMP. We show that the overall structure of the TRIP8b bound conformation of the CNBD closely resembles the apo state. In addition, we use DEER between the CNBD and TRIP8bcore and nuclear magnetic resonance (NMR) to localize the binding site for TRIP8b on the CNBD. Finally, to understand the mechanism of TRIP8b inhibition of cAMP regulation of HCN channels, we performed binding studies of cAMP and TRIP8b on the CNBD and developed a multi-state mathematical model to explore 1) whether cAMP and TRIP8b can bind simultaneously to HCN channels, 2) whether TRIP8b reduces the affinity for cAMP binding, and 3) whether TRIP8b reduces the effect of cAMP on HCN channels by preventing the structural rearrangements associated with channel opening. 1750-Plat Epilepsy Related Slack Channel Mutants Lead to Channel Over-Activity by Two Different Mechanisms Qiong-Yao Tang1, Fei-Fei Zhang1, Jie Xu1, Ran Wang2, Jian Chen2, Zhe Zhang1. 1 Jiangsu Key Labratory of Anesthesiology, Jiangsu Province Key Laboratory of Anesthesia and Analgesia, Xuzhou Medical College, Xuzhou, China, 2 Anesthesiology Department, Xuzhou Medical College, Xuzhou, China. So far, 12 sodium-activated potassium channel (KCNT1, Slack) genetic mutants had been found in severe early-onset epilepsy patients by whole genome sequence. However, the biophysical properties change of these mutated Slack channels and the underlying mechanisms had not been fully investigated. In this abstract, we first measured the KD value of sodium sensitivity of these mutant channels by inside-out patch. We found that two mutants (R409Q and R455H) on the RCK1 domain and one mutant (Y775H) on the RCK2 domain decreased the KD value of sodium sensitivity. Furthermore, we found that the double mutant (R455H/R409Q) on RCK1 domain can decrease KD value of sodium sensitivity further, reflecting these mutants allosterically regulated sodium affinity of Slack channel. In addition, electrophysiology and molecular simulation reflected the mutant Y775H may directly facilitate sodium binding of Slack channel. Second, single channel recording data revealed that all mutants lead to over-activity of Slack channel even if they did not change the sodium sensitivity. We set up a two-step activation model to explain the gating mechanism change and concluded that these mutant channels lead to channel over-activity can be categorized as two different mechanisms. 1751-Plat Quantum Calculations Show a Water Column in a Potassium Ion Channel Pore, and its Role in Gating and Conduction Alisher M. Kariev, Michael E. Green. Chemistry, City College of the City Univ of NY, New York, NY, USA. A continuous water column is shown to exist in the pore of a potassium channel. Quantum calculations (HF/6-31G*) were performed on the Kv1.2 channel (pdb:2A79/3Lut), giving the structure of the pore with 50 water molecules. Calculations were done with 0, 1, or 2 ions. The protein from the PVPV intracellular gating region to the entrance to the selectivity filter, including the ˚ ) was included, with water extyrosine of the TVGYG sequence (z14.1 A tending slightly past the region. Results show how the ion(s) restructure(s) Tuesday, February 10, 2015 the water; water structure largely controls interaction of ions with one another and with the protein. We had previously posited that there must be an oscillating gate including water, which changes structure with ion position (Int’l J. Molec. Struct., 2012, 13, 1680); newly calculated structures correspond to the previously hypothetical states. These results also indicate the presence of two other local minima for the ion, one in the cavity, one at the entrance to the selectivity filter, where the protein replaces water in solvating the ion. A single ion there is sufficiently distant that the gate water cluster returns to its original form. There is some charge transfer to the ion from unshared electron pairs on oxygen. Finally, we determine the water cluster structure’s relation to boundary protein. The water column in the pore is continuous, and energy barriers for an ion moving up in the pore appear compatible with observed conductivity in the channel. Thus, interactions between ions are largely modulated by water structure interacting with protein structure, not simply electrostatics. 1752-Plat PIP2 and Surface Expression Underlie Apo-Calmodulin Dependent Kv7.2/ KCNQ2 Current Potentiation Carolina Gomis-Perez1, Maria Virginia Soldovieri2, Araitz Alberdi1, Paolo Ambrosino2, Michela Di Maria2, Alessandro Alaimo1, Ganeko Bernardo-Seisdedos1, Covadonga Malo1, Pilar Areso3, Maurizio Taglialatela2,4, Alvaro Villarroel1. 1 Unidad de Biofisica CSIC,UPV/EHU, Leioa, Spain, 2Dept. of Medicine and Health Science, University of Molise, Campobasso, Italy, 3Dept. of Pharmacology, UPV/EHU, Leioa, Spain, 4Dept. of Neuroscience, University of Naples ‘‘Federico II’’, Naples, Italy. Calmodulin (CaM) availability regulates neuronal excitability by controlling the functional density of the potassium voltage-dependent M-current, but the basis of this fundamental process is not well understood. We have examined the consequences of varying CaM levels on the main constituents of the M-current, which are Kv7.2 and Kv7.3 subunits. We found that CaM increased the functional density of Kv7.2 channels but not that of Kv7.2/3 or Kv7.3. Similar effects were also observed with a mutant CaM unable to bind Ca2þ. In agreement with the proposed role in trafficking, CaM augmented the number of channels at the plasma membrane. To address the involvement of PIP2, its abundance was transiently reduced by activating Danio rerio voltagesensitive phosphatase (Dr-VSP). CaM-dependent Kv7.2 current potentiation was linked to increased resistance to PIP2 depletion. In contrast, sequestration of endogenous CaM led to a reduction in the functional current density for the three Kv7 channel combinations examined (2, 2/3 and 3). These results suggest that CaM is affecting two mechanisms: one leads to an increase in the number of channels at the plasma membrane, and the second involves a PIP2-dependent increase in opening probability 1753-Plat The Mechanism of KCNE1 Modulation of KCNQ1 Channels Mark A. Zaydman1, Marina Kasimova2, Kelli Delaloye1, Jingyi Shi1, Hongwu Liang1, Zachary Beller1, Mounir Tarek2, Jianmin Cui1. 1 Biomedical Engineering, Washington University in St. Louis, St. Louis, MO, USA, 2Universite´ de Lorraine, Nancy, France. The IKs current controls action potential duration in the heart, and abnormal function of this current causes cardiac arrhythmias. The IKs current is carried by the voltage activated KCNQ1 potassium channel associated with KCNE1 b-subunits. 18 years of study have shown that KCNE1 drastically modulates every characteristic of KCNQ1, such that it would appear as if KCNQ1 and KCNQ1þ KCNE1 were completely unrelated channels. However, no coherent mechanism has been provided that can explain all these drastic changes, which are essential for the physiological role of IKs. Here we show that KCNE1 alters the state-dependent interactions (coupling) between the voltage-sensing and pore-gate domains of KCNQ1 and that this sole mechanism is sufficient to explain all these changes. Contrary to conventional belief that the voltage-sensing domain must reach the fully-activated state before promoting pore-opening, we found that the KCNQ1 channels can open when the voltage-sensing domain is at intermediate and fully-activated states. Importantly, the intermediate-open and activated-open channels differ in voltage-dependence, ion-permeation, pharmacology and dependence on PIP2, a cofactor for coupling between the voltage-sensing and pore-gate domains. By changing the coupling, KCNE1 prevents the intermediate-open state and changes the properties of the activated-open state, thereby bringing about the characteristics of the IKs current. These results indicate that, during voltage-dependent ion channel gating, every-state of the voltage-sensing 349a domain along its activation pathway is coupled to the conformation of the pore domain through a unique set of protein-protein and protein-lipid interactions. These interactions determine both the open-probability and the open-pore properties. 1754-Plat Disruption of Assembly/Calmodulin-Binding Coupling and CalmodulinDependent Potentiation of Kv7.2 Channels by a Epileptogenic Helix D Mutation Araitz Alberdi1, Ganeko Bernardo-Seisdedos1, Carolina Gomis-Perez1, Alessandro Alaimo1, Covadonga Malo1, Elisabeth Butz2, Christian Wahl-Schott2, Pilar Areso3, Alvaro Villarroel1. 1 Unidad de Biofı´sica CSIC, UPV/EHU, Leioa, Spain, 2Zentrum fu¨r Pharmaforschung - Ludwig-Maximilians-Universita¨t Mu¨nchen, Munich, Germany, 3Pharmacology Department, UPV-EHU, Leioa, Spain. Kv7.2 is a main component of the neuronal M-current, whose density depends on the interaction with calmodulin (CaM). Several observations hint that CaM binding is coupled to Kv7 subunit multimerization, but such important process is not well-understood. Whereas subunit specific assembly of Kv7 channels depends on helix D, CaM binds to helices A and B located 60 residues upstream. By studying the consequences of the helix D L609R mutation found in patients suffering Neonatal Familial Seizures, we confirm that helix D is crucial for C-terminal multimerization. Surprisingly, disruption of helix D-dependent C-terminal multimerization by L609R has no major consequences for Kv7.2 channel electrophysiological properties. FRET results suggest that CaM enhances multimerization of the wt C-terminal domain, but has marginal effects when the helix D mutation is present. The L609R mutation weakens the interaction with CaM and disrupts CaM-dependent Kv7.2 potentiation. These results underscore a complex interconnection between CaM, PIP2 and multimerization on the regulation of homomeric Kv7.2 channels. 1755-Plat Statin Inhibits IKs Internalization in Response to Prolonged Stress Stimulus Xiaorong Xu Parks1, Elsa Ronzier1, Rachael E. Abraham2, Jin O-Uchi1, Coeli M. Lopes1. 1 CVRI, University of Rochester, Rochester, NY, USA, 2Biology, University of Rochester, Rochester, NY, USA. Statins are among the most commonly prescribed drug classes, and their use is expected to increase due to recent changes in therapy guidelines. Although statins have been suggested to shorten QT interval in heart failure patients, the effect of this drug class on cardiac electrophysiology has been incompletely studied. Here we study the effect of statins on one of the major repolarizing currents in the heart, IKs. We show that in response to a known stress stimulus (sustained alpha-AR stimulation), IKs undergoes clathrin-dependent internalization. Statin treatment inhibits this internalization. Our results suggest that statin regulates IKs internalization by inhibiting activation of Rab5, a GTPase involved in endocytic pathway regulation. We observe no significant effect of statin treatment on unstimulated IKs function and acute receptor regulation. Our results suggest that statins may prevent pathological QT prolongation in heart failure patients by inhibiting pathological remodeling and IKs internalization, while maintaining physiological trafficking and function of the channel. Platform: Membrane Receptors and Signal Transduction 1756-Plat Confrontational Dynamics of a GPCR Revealed by Single Molecule FRET Reza Vafabakhsh1, Joshua Levitz2, Ehud Y. Isacoff1. 1 Molecular and Cell Biology, University of California at Berkeley, Berkeley, CA, USA, 2Biophysics Graduate Group, University of California at Berkeley, Berkeley, CA, USA. G protein-coupled receptors (GPCRs) function as sensory, synaptic and hormone receptors. Metabotropic glutamate receptors (mGluRs) are dimeric class C GPCRs that mediate synaptic transmission and plasticity, and serve as drug targets for Parkinson’s disease, schizophrenia and autism. We developed an assay based on single molecule Forster resonance energy transfer (smFRET) to probe the conformational dynamics of full length mammalian group II mGluRs. Our assay revealed the structural dynamics associated with the ligand 350a Tuesday, February 10, 2015 binding and provided a dynamical basis for the efficacy of partial agonists or full agonists, as well as the modulation of activation by positive allosteric modulators (PAMs). Finally, our work provides a general model for the activation process of mGluRs. 1757-Plat Biased Agonism at Opioid Receptors: Insights from Analysis of Structural Interaction Fingerprints Davide Provasi, Paola Bisignano, Marta Filizola. Icahn School of Medicine at Mount Sinai, New York, NY, USA. Biased agonism, that is the ligand-dependent selectivity for specific signaling pathways in a G protein coupled receptor (GPCR), is an emerging, promising strategy for the development of safer drugs. In the case of the kappa opioid receptor (KOPr), its preferential activation of G-protein versus b-arrestin has been proposed to provide a more direct route to discovering non-addictive opioid therapeutics with reduced side effects. In fact, the KOPr-mediated dysphoria that usually accompanies the beneficial analgesic effect of KOPr agonists has recently been attributed to the activation of the p38 mitogenactivated protein kinase pathway, which is believed to follow arrestin recruitment to the activated KOPr. This observation suggests that KOPr agonists that selectively activate the G protein, but do not recruit arrestin, may be more effective analgesics since they would not exhibit the adverse effects triggered by the arrestin pathway. Based on the above, understanding the structural and chemical determinants of biased agonism at the KOPr is highly desirable as it can guide the discovery/ design of improved therapeutics. Here, we employ flexible docking of a set of recently characterized functionally selective KOPr ligands to establish a predictive model for G protein-biased agonism at this receptor based on characteristics of modes of interaction between the ligand and the KOPr. We apply the resulting classifier to a large set of established KOPr agonists to assess their ability to preferentially promote G protein coupling, arrestin recruitment, or both. 1758-Plat Conformational Dynamics of a G Protein-Coupled Receptor at the SingleMolecule Level Rajan Lamichhane, Jeffrey J. Liu, Raymond C. Stevens, David P. Millar. Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, USA. G protein-coupled receptors (GPCRs) are the integral membrane proteins that detect extracellular ligands and mediate signal transduction. Binding of ligands promotes transitions from inactive to active receptor conformations, which are recognized by intracellular effectors. Chemically distinct ligands may trigger different signaling responses by altering dynamics of the receptor. We developed a single-molecule fluorescence system to observe conformational switching of the b2-adrenergic receptor in real-time within a native-like membrane environment. The receptor was covalently labeled with a photostable and environmentally-responsive Cy3 fluorophore at the cytoplasmic end of either transmembrane (TM) helix VI or helix VII. Individual receptor molecules were incorporated in phospholipid nanodiscs, tethered to a microscope cover slip and visualized over time by total internal reflection fluorescence microscopy. We observed spontaneous transitions of TM helices VI and VII between inactive and active conformations, even in the absence of any ligands. Studies of ligands that span a comprehensive range of pharmacological efficacies showed that full agonists shorten the time spent in the inactive conformations and prolong the time in the active conformations, leading to an increased population of active species, while an inverse agonist prolonged the time spent in inactive conformations. These observations provide new insights into the mechanism of GPCR activation and the molecular basis for the variable pharmacological efficacies of different drug molecules. 1759-Plat Entry from the Lipid Bilayer: A Novel Pathway for Inhibition of a Peptide G-Protein Coupled Receptor by a Lipophilic Small Molecule Michael P. Bokoch1,2, Hyunil I. Jo1, James R. Valcourt3, Yoga Srinivasan1, Kazuma Yasuhara1,4, Albert C. Pan3, Ron O. Dror3, David E. Shaw3,5, William F. DeGrado1, Shaun R. Coughlin1. 1 Cardiovascular Research Institute, University of California, San Francisco, San Francisco, CA, USA, 2Anesthesia and Perioperative Care, University of California, San Francisco, San Francisco, CA, USA, 3D.E. Shaw Research, New York, NY, USA, 4Graduate School of Materials Science, Nara Institute of Science and Technology, Nara, Japan, 5Biochemistry and Molecular Biophysics, Columbia University, New York, NY, USA. G-protein-coupled receptor (GPCR) crystal structures reveal exquisite details of interactions between amino acids and bound ligands. However, little is known about the pathways by which ligands enter their binding sites. Binding pathways are essential determinants of association and dissociation rates and thus underlie kinetic selectivity, a phenomenon in which a ligand primarily affects a particular receptor despite having similar binding affinity for several closely related receptors. Protease-activated receptor 1 (PAR1) is a GPCR whose endogenous agonist is a tethered, water-soluble peptide generated by thrombin cleavage. By contrast, the PAR1-specific antagonist vorapaxar is a lipophilic small molecule that binds in a pocket almost entirely occluded from extracellular solvent. Based on temperature-accelerated molecular dynamics simulations, we hypothesize that vorapaxar enters the PAR1 binding pocket from the lipid bilayer between transmembrane helices 6 and 7 (TM6 and TM7). To test this hypothesis, we take a chemical biology approach and synthesize vorapaxar derivatives with alkyl chains extending along the predicted binding pathway. In cell signaling assays, we find that the on-rate for vorapaxar derivatives with increased bulk between TM6 and TM7 is no slower than that of vorapaxar. These data indicate that vorapaxar likely enters the binding pocket from the lipid bilayer, through the cleft between TM6 and TM7. While other groups have reported experimental evidence that retinal binds to rhodopsin through a channel from the lipid bilayer, our study provides the first such data for a ligand binding to a peptide GPCR. Membrane drug entry mechanisms, such as the pathway we describe for vorapaxar binding to PAR1, may be important for understanding kinetic selectivity of lipophilic GPCR ligands and may inform selective drug design. 1760-Plat Single Molecule Imaging of M2 Muscarinic Receptors in Live Heart Explants Gregory I. Mashanov1, Tatiana A. Nenasheva1, Ross A. Breckenridge2, Nigel J.M. Birdsall1, Justin E. Molloy1. 1 Physical Biochemistry, MRC National Institiute for Medical Research, London, United Kingdom, 2MRC National Institiute for Medical Research, London, United Kingdom. TIRF microscopy allows us to visualize individual fluorophores in living cells. We have developed automated algorithms to detect and track thousands of individual molecules and have studied GPCRs (G-protein coupled receptors) tagged with fluorescent ligands or fluorescent proteins in live cultured cells and freshly dissected tissue slices. We have found that M1 and M2 muscarinic receptors diffuse freely at the plasma membrane with their mobility dependent upon both cell-type and temperature. The mobility of M2 receptors tagged with the fluorescent antagonist (Cy3B-telenzepine) matches the mobility of M2-GFP molecules imaged under the same conditions. Fluorophore tracking allowed direct observation of transient homo-dimerisation of M1 and M2 receptors which we confirmed using two-color imaging. At a membrane receptor density of 2 mm2, 30% of receptors were dimeric and the dimer lifetime was ~1 s1. We have found that only ~10% of isolated primary cardiomyocytes and cells in the mice heart slices expressed M2 receptors on the detectable level. The receptors undergo unrestricted diffusion and therefore were evenly distributed across the cell membrane. The M2 density in freshly isolated embryonic cardiomyocytes was ~1 mm2, increasing at birth to ~3 mm2 and decreasing back to ~1 mm2 after birth. Whilst some M2 receptors formed reversible dimers the majority were monomeric. The receptor mobility was approximately 4-times faster in freshly dissected heart slices (0.6 mm2 s1) than in cultured primary cardiomyocytes. Knowing receptor mobility and density we used Monte Carlo simulations to estimate an encounter rate of 5-10 collisions per second. This may explain the observed electrophysiological latency between the application of acetylcholine and GIRK channel opening. 1761-Plat The Structural Basis for Lipid a Recognition in the CD14 Innate Immune Co-Receptor Nils A. Berglund1,2, Daniel A. Holdbrook1, Syma Khalid2, Peter J. Bond1,3. 1 Bioinformatics Institute, Agency for Science, Technology and Research, Singapore, Singapore, 2Chemistry, University of Southampton, Southampton, United Kingdom, 3Department of Biological Sciences, National University of Singapore, Singapore, Singapore. The CD14 co-receptor is specialized for recognition of bacterial lipopolysaccharide (LPS). On the surface of macrophages and other immune cells, it transfers LPS and its bioactive component lipid A to the MD-2 protein in complex with Toll-Like Receptor 4 (TLR4), and is hence crucial in activating the innate immune system via the TLR4 signalling pathway. In the case of severe infections, lipid A can cause sepsis through over-activation of the immune response, leading to multiple organ failure and death, and has become a major target for anti-septic drugs. Unfortunately, the mechanism by which lipid A is transferred to CD14, and the detailed mode(s) of associated binding, are Tuesday, February 10, 2015 unknown. In this study we have used atomically detailed molecular dynamics simulation approaches to uncover the mechanism by which lipid A is transferred to, interacts with and binds to a hypothesized amino-terminal pocket in CD14. We modelled the interactions and dynamics of CD14 in the presence of a range of lipid ligands, including control fatty acid systems, and lipid A in monomeric and aggregate/micelle forms. These simulations were run in order to observe the spontaneous ligand binding process, and have subsequently been extended to establish the thermodynamics of ligand recognition. Our results emphasise the dynamic nature of the amino-terminal pocket which allows it to adapt its volume to widely varying ligand size, consistent with the broad specificity of CD14. We have also identified a possible ligand gating mechanism consistent with available NMR data, and key sites that may be essential for LPS/lipid A binding which may ultimately be targeted by novel anti-septic drugs. 1762-Plat Inside-Out Signaling of Oncogenic EGFR Mutants Promotes LigandIndependent Dimerization Christopher C. Valley1, Donna J. Arndt-Jovin2, Thomas M. Jovin2, Mara P. Steinkamp1, Alexey I. Chizhik3, Narain Karedla3, William S. Hlavacek4, Bridget S. Wilson1, Keith A. Lidke5, Diane S. Lidke1. 1 Department of Pathology, University of New Mexico, Albuquerque, NM, USA, 2Laboratory of Cellular Dynamics, Max Planck Institute for Biophysical Chemistry, Go¨ttingen, Germany, 3III. Institute of Physics, Georg August University, Go¨ttingen, Germany, 4Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM, USA, 5 Department of Physics and Astronomy, University of New Mexico, Albuquerque, NM, USA. Mutations within the epidermal growth factor receptor (EGFR/erbB1/Her1) are often associated with carcinogenesis. Specific mutations common in non-small cell lung cancer (NSCLC), including EGFR-L858R and EGFRDL747-P753, lead to ligand-independent phosphorylation, however the molecular mechanism by which these mutations in the EGFR kinase domain confer constitutive activity remain unknown. Here, using multiple sub-diffractionlimit imaging modalities, we reveal the altered behavior of NSCLCassociated EGFR mutants within the plasma membrane_including altered receptor dimerization, dynamics, and structure_which collectively dysregulate receptor activity. Using multi-color single particle tracking (SPT) and Hidden Markov Model analysis, EGFR mutants are shown to form stable dimers in the absence of ligand and exhibit a slower mobility that is consistent with receptor signaling. These results were confirmed using two-color, single-molecule super-resolution microscopy (dSTORM) to visualize the spatial distribution of receptors. Receptor clustering was quantified by localization-based crosscorrelation analysis to show ligand-induced aggregation of EGFR as well as ligand-independent aggregation of EGFR mutants. Since the receptor ectodomain is known to play a critical role in dimerization, live cell FRET measurements between the EGFR N-terminus and the plasma membrane was used to quantify changes in ectodomain structure. We found that unliganded EGFR mutants are more readily found in the extended conformation, similar to the ligand bound wild type receptor. Therefore, mutation within the kinase domain biases the structural equilibrium of the extracellular domain toward a dimercompetent state. Collectively, these data support a model where oncogenic signaling from NSCLC-associated EGFR mutants is a result of productive dimerization between non-ligand bound receptors. Furthermore, because these mutations are found in the kinase domain, this work introduces the concept that oncogenic EGFR signaling may be controlled in part by a form of ‘‘inside-out’’ signaling. 1763-Plat Mechanisms of Autoinhibition and Dimerization of the EGF Receptor Family Patrick Byrne1, Kalina Hristova2, Daniel Leahy1. 1 Biophysics and Biophysical Chemistry, Johns Hopkins University, Baltimore, MD, USA, 2Materials Science and Engineering, Johns Hopkins University, Baltimore, MD, USA. The epidermal growth factor receptor (EGFR/ErbB) family of receptor tyrosine kinases initiates cell signaling in response to growth factors, thereby regulating cell growth, migration and division. Four members comprise this family (EGFR/HER1/ErbB1, ErbB2, ErbB3 and ErbB4), and activating mutations in each protein are associated with severe cancers of the lung, colon, head and neck. ErbB proteins consist of an extracellular ligand binding domain, a single transmembrane alpha-helix, and an intracellular kinase domain. Structural and functional studies of isolated domains of the EGFR family have revealed details of the complex mechanism of receptor regulation. Somewhat lacking, however, is 351a our knowledge of the full-length receptor in its native membrane environment. Ligand binding is known to promote receptor dimerization and activation, but it is unclear how receptor behavior is altered by the two-dimensional environment of the plasma membrane. In particular, the mechanism by which receptor domains cooperate to transmit signals across the membrane is not well understood. Toward this end, we employed quantitative FRET microscopy to study dimerization of EGFR family proteins in plasma membrane-derived vesicles. This method measures FRET efficiency as a function of two-dimensional receptor concentration in the membrane. We investigated four receptor pairs: homodimers of ErbB1, ErbB2, and ErbB3, as well as heterodimers containing ErbB2 and ErbB3. We first observe that the isolated transmembrane (TM) domains have an intrinsic propensity to associate. Next, we demonstrate that the extracellular domains function to prevent TM domain interactions. Finally, we show that the intracellular domains alone are sufficient to drive receptor dimerization in the absence of ligand. Dimerization occurs in a concentration-dependent manner, and ligand shifts this threshold to low receptor concentration. Importantly, both of these dimerization events occur over a physiologically relevant concentration range. Platform: DNA Structure 1764-Plat Solid-To-Fluid DNA Transition Inside HSV-1 Capsid Close to the Temperature of Infection Alex Evilevitch1, Udom Sae-Ueng1, Dong Li1, Xiaobing Zuo2, Jamie Huffman3, Fred Homa3, Donald Rau4. 1 Physics, Carnegie Mellon University, Pittsburgh, PA, USA, 2Argonne National Laboratory, Argonne, IL, USA, 3School of Medicine, Univ. of Pittsburgh, Pittsburgh, PA, USA, 4National Institute of Health, Bethesda, MD, USA. DNA in the human Herpes simplex virus type 1 (HSV-1) capsid is packaged to a tight density. This leads to tens of atmospheres of internal pressure responsible for the delivery of the herpes genome into the cell nucleus. In this study we show that despite its liquid crystalline state inside the capsid, the DNA is fluid-like which facilitates its ejection into the cell nucleus during infection. We found that the sliding friction between closely packaged DNA strands, caused by interstrand repulsive interactions, is reduced by the ionic environment, mimicking that of epithelial cells and neurons susceptible to herpes infection. However, variations in the ionic conditions corresponding to neuronal activity can restrict DNA mobility in the capsid, making it more solid-like. This can inhibit intranuclear DNA release and interfere with viral replication. In addition, the temperature of the human host (37 C) induces a disordering transition of the encapsidated herpes genome which reduces interstrand interactions and provides genome mobility required for infection. 1765-Plat Nanopore Sensors for Analysis of Circular DNA Topology Eric Krueger1,2, Jiwook Shim3,4, A. Nicole Chang1, Basheer Subei5, Arman Fathizadeh5, Katie Livingston1, Paul Davis1, Elton Graugnard1, Fatemeh Khalili-Araghi5, Rashid Bashir3, David Estrada1, Daniel Fologea2. 1 Materials Science and Engineering, Boise State University, Boise, ID, USA, 2 Physics, Boise State University, Boise, ID, USA, 3Bioengineering, University of Illinois at Urbana - Champaign, Urbana, IL, USA, 4Micro and Nanotechnology Laboratory, University of Illinois at Urbana - Champaign, Urbana, IL, USA, 5Physics, University of Illinois at Chicago, Chicago, IL, USA. Over the course of its life cycle, a cell’s DNA undergoes many carefully orchestrated topological changes, which facilitate vital cellular processes such as replication and transcription. Consequently, unresolved conformational defects in the structure can interfere with critical interactions, and may result in genetic anomalies that culminate in cell death. As a result, DNA has become the target for numerous anti-cancer treatments which seek to induce structural changes to inhibit tumor growth [1, 2]. The development of innovative anti-cancer treatments can be greatly enhanced by highthroughput low-cost methods to characterize their effects on DNA topology [3]. In this study, we use Si3N4 solid-state nanopores to investigate changes in circular DNA topology induced by intercalation of ethidium bromide (EtBr). Our measurements reveal three distinct current blockade levels and a six-fold increase in translocation times for EtBr treated circular DNA as compared to untreated circular DNA. We attribute these increases to changes in the supercoiled topological state hypothesized to be branched or looped structures formed in the circular DNA molecule. Further evidence of the conformational changes is demonstrated by qualitative atomic force 352a Tuesday, February 10, 2015 microscopy analysis. We evaluate our experimental data correlating topological changes with specific translocation event morphologies using all-atom Molecular Dynamics simulations. Our results provide new fundamental insight into characterizing DNA topology, and has important implications for anticancer drug treatment and design. 1. K. Gurova, Future Oncology 5, 1685 (2009). 2. R. Martinez and L. Chacon-Garcia, Current Medicinal Chemistry 12, 127 (2005). 3. R. Palchaudhuri and P. J. Hergenrother, Current Opinions in Biotechnology 18, 497 (2007). 1766-Plat Mechanical Properties and Strand Invasion of Duplex Telomere DNA Probed using Magnetic Tweezers Xi Long, Michael D. Stone. Chemistry and Biochemistry, UC Santa Cruz, Santa Cruz, CA, USA. Telomeres are specialized chromatin structures that protect chromosome ends from nucleolytic processing by DNA repair machinery. The foundation of human telomere structure consists of a long array of tandem duplex DNA sequences (TTAGGG) and terminates with a single-stranded 3’ end. To protect the chromosome end, telomeres are thought to adopt a lariat structure known as a telomere-loop (T-loop)1. T-loops are stabilized by DNA displacement loops (D-loops) generated by the invasion of a single-stranded telomeric DNA tail into an adjacent region of duplex telomere. Recent studies suggest that telomere-associated proteins promote strand invasion during telomeric D-loop formation through the application of torque to the DNA2. Although the molecular mechanism of T-loop formation has been described using biochemical approaches, the torque response and internal structural equilibrium of duplex telomeric DNA are not well characterized. To probe the mechanical properties of duplex telomeric DNA, we developed a magnetic tweezers assay to detect the response of single telomeric DNA molecules to precisely applied degrees of tension and torque. Rotation-extension curves under varying tension demonstrate that the repetitive telomere DNA sequence is more refractory to torque-induced denaturation than a non-telomeric control molecule of comparable GC content. In addition, force-extension analysis of negatively supercoiled telomeric DNA in the presence of different counter-ions (Kþ vs. Liþ), reveals that transient torque-induced denaturation of duplex telomeric DNA promotes a structural transition into stable DNA G-quadruplexes. Lastly, using a single molecule DNA topology-based assay, we directly monitor the torquedependent invasion of single stranded telomere DNA primers into duplex telomeric DNA tethers. Our results provide insight into the molecular mechanisms of telomere-associated proteins and enzymes during structural remodeling of telomeres. 1. Griffith JD, et al. Cell 97: 503-514. 2. Amiard S, et al. Nat Struct Mol Biol 14:147-154. 1767-Plat High-Throughput Quantification of the Impact of Different Osmolytes on the Thermal Stability of DNA Prem K. Sinha, Mikhail Sinev, Jo¨rg Ro¨sgen. Biochemistry & Molecular Biology, Pennsylvania State University, Hershey, PA, USA. Small molecules (osmolytes) are known to either stabilize or destabilize proteins/nucleotides depending on the concentrations and/or solvent conditions. The presence of different molecules and ions in the surrounding medium affects the stability of DNA in solution. In this work, we have developed a HighThroughput method for quantifying the energetic impact of addition of various osmolytes on short DNA duplexes. Six 19-base pair, non-self-complementary duplex DNA oligomers along with a 16-base pair control duplex DNA, having varied GC-content (ranging from 16% to 79%), nearest neighbors and end sequences were used. We sampled thirteen different osmolytes that are common in humans and throughout nature by covering different chemical classes including, sugars, polyols, amino acids, and methylamines. Varying concentrations of these osmolytes (from 0.5 M up to 3.0 M) were examined for their effects on these duplexes. Experiments were performed in 384-well plates that were prepared using a robotic device, which was calibrated for the correct dispense volume for different components of the plate. Temperature-induced melting transitions monitored by fluorescence were measured for these duplexes and the Tm values along with the m-values and melting transition enthalpies were determined. Conventional approaches, including Circular Dichroism (CD) were used to verify the thermodynamic parameters. Osmolytes had varied effects on DNA stability, and the (de)stabilizing effect does not necessarily correlate with their effects on proteins. The m-values can drastically depend on the GC-content. 1768-Plat Real Time Transposable Element Dynamics Thomas E. Kuhlman. Physics, University of Illinois at Urbana-Champaign, Urbana, IL, USA. Transposable elements are mobile genetic elements that are capable of selfcatalyzed excision or copying from their host’s genome, followed by integration back into another location within the genome. Transposable elements increase in number over time as a result of this activity, and as a consequence can make up a substantial portion of the host’s genome as ‘‘junk DNA’’; both active and dormant transposable elements make up at least 45% of the human genome, and up to 85% of the maize genome. Additionally, the unpredictable reintegration of transposable elements into coding or control regions of the host’s genome can have dramatic effects on gene expression, and, as a result of this inherently mutagenic nature, transposable elements are thought to be a major source of genome plasticity driving evolution and are implicated as the direct causative agents of many human diseases, including hemophilia, porphyria, severe combined immunodeficiency, muscular dystrophy, and breast and colon cancers. Despite their ubiquity and potential importance, very little is currently known about the dynamics of transposon propagation through genomes, and their contribution to evolution is inferred from comparative analyses of the genome sequences of related organisms. In this talk I will describe methods developed by my lab employing fluorescent microscopy, microfluidic, and molecular biology techniques to allow the direct visualization of transposable element activity in single cells and in real time. The proposed experimental system is extensible to all types of transposable elements and all cell types, from bacteria to human. 1769-Plat Effect of Methylation on the Nanomechanics of Double-Stranded DNA Csaba I. Pongor1, Pasquale Bianco2, Miklo´s Kellermayer1. 1 Department of Biophysics, Semmelweis University, Budapest, Hungary, 2 Department of Physics, University of Florence, Florence, Italy. In its physiological environment DNA is constantly exposed to mechanical stress. The nanomechanical properties of DNA influence not only its response to stress but also its interaction with proteins. Despite its crucial role in epigenetics, little is known about how methylation affects the nanomechanical properties of DNA. To investigate the impact of methylation on DNA nanomechanics, here we manipulated single molecules of chemically or enzymatically methylated DNA and compared their properties with those of nonmethylated DNA. As a model we used a 3312-base-pair long sequence of lambda-phage DNA that met the criteria of a CpG island. Chemically methylated DNA was prepared with PCR containing 5-methyl-CTP in the reaction mixture. For enzymatic methylation the M.Sss.I methyltransferase was used. Single DNA molecules were mechanically manipulated with force-measuring optical tweezers in repeated stretch-relaxation cycles. Surface-adsorbed DNA molecules were studied by using atomic force microscopy (AFM). We found that the molecular contour length, bending rigidity and intrinsic stiffness were decreased in methylated DNA, pointing at structural and nanomechanical alterations. Furthermore, the cooperative overstretch transition was significantly longer in the methylated form of the molecule, suggesting that the dynamics of intramolecular rearrangements were also affected. AFM measurements of DNA molecules adsorbed to mica surface substantiated the significant reduction of molecular contour length in methylated DNA. By contrast, the apparent bending rigidity of the surface-adsorbed methylated DNA was increased, which is most likely caused by interactions between DNA and the mica surface. In sum, methylation leads to an axial compaction of the dsDNA structure, an increase in bending flexibility in the low-force regime and an increase in axial compliance at higher forces (>20pN). Conceivably, modulation of DNA structure and nanomechanics caused by methylation leads to a complex control of structural accessibility and association kinetics of DNA-binding proteins. 1770-Plat Thermodynamics for the Interaction of PEG-PLL Copolymers with DNA Hui-Ting Lee, Alexander J. Lushnikov, Irine Khustsishvili, Luis A. Marky. Pharmaceutical Sciences, University of Nebraska Medical center, Omaha, NE, USA. One focus of our research is to select polycations to deliver oligonucleotides into the cell for the control of gene expression. In this work, we report on the interaction of poly(ethylene glycol)-b-poly-L-lysine (PEG-PLL) copolymer with a variety of DNA molecules. Specifically, three PEG-PLL copolymers with similar PEG segment but different length of the PLL chains were used to interact with DNA duplexes as a function of duplex length. A combination Tuesday, February 10, 2015 of spectroscopic and calorimetric techniques was used to investigate the unfolding of both DNA molecules and polycation-DNA complexes, and to determine their thermodynamic binding profiles. The resulting polycation-DNA complexes were stable in aqueous solution at room temperature. The binding of each copolymer to DNA stabilized the helix-coil transition of all DNA molecules, yielding binding affinities of ~104 M-1, which were lowered by the increase in salt concentration. However, binding affinities of 105 were obtained with the ethhidium bromide displacement essay. Isothermal titration calorimetric experiments yielded negligible heats of interaction. Therefore, the favorable formation of the copolymer-DNA complexes is entropy driven which was rationalized in terms of the release of both counterions and water molecules upon complex formation. In summary, polycation binding to DNA was found to be electrostatic in nature, i.e., the positively charged lysine groups formed ion pairs with the negatively charged phosphate groups of DNA. Supported by Grant MCB-1122029 from the National Science Foundation. 1771-Plat Correlating Drug Binding Affinities with Base Pair Opening Rates in DNA Mary E. Hatcher1, Mary Creedon2. 1 Keck Science Department, The Claremont Colleges, Claremont, CA, USA, 2 Keck Science Department, Scripps College, Claremont, CA, USA. The cyclic AMP responsive element (Cre) is a highly conserved stretch of DNA that is involved in the activation of gene transcription. Our group has studied the drug binding of a fluorescent derivative of a DNA intercalating anti-cancer drug, 7-amino actinomycin D (7-AMD), to the Cre sequence in different sequence contexts. We initially analyzed the DNA backbone conformation of Cre samples with varying flanking sequences and correlated these values to the binding affinities of 7-AMD. These studies revealed several anomalies that suggest that the conformation described by BI/BII content of the DNA backbone is, at most, only partially responsible for the 7-AMD binding affinity to the Cre sequence. This result is not surprising as 7AMD has a conjugated ring structure in addition to its peptidyl side chains which interact with the backbone. As a result, we began studying DNA base pair opening rates and correlating these with 7-AMD binding affinities to account for the intercalation of 7-AMD into the Cre sequence. DNA base pair opening rates were determined by tracking imino proton exchange via NMR spectroscopy in the presence of a varying concentration of base catalyst. In this study, both two-dimensional NOESY and one-dimensional H1 NMR spectroscopy are used to track the change in line widths of the imino protons of the central Cre binding site for five sequences with varying flanking sequences. Trends in opening rates reveal that sequences with strong 7-AMD binding feature slower base pair opening dynamics than sequences with weaker 7-AMD binding. These results suggest that local base stacking is important for 7-AMD binding, a hypothesis we are currently investigating with UV spectroscopy. Platform: Exocytosis, Endocytosis, and Membrane Fusion 1772-Plat Microtubule Motors Drive Plasma Membrane Tubulation in ClathrinIndependent Endocytosis Charles A. Day1, Nicholas W. Baetz1, Ajit Tiwari1, Kimberly R. Drake1, Courtney A. Copeland1, Lewis J. Kraft1, Bing Han1, Daniel J. Chinnapen2, Michael W. Davidson3, Randall K. Holmes4, Michael G. Jobling4, Trina A. Schroer5, Wayne I. Lencer2, Anne K. Kenworthy1. 1 Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN, USA, 2GI Cell Biology, Children’s Hospital, Boston, MA, USA, 3 National High Magnetic Field Laboratory, The Florida State University, Tallahassee, FL, USA, 4Department of Microbiology, University of Colorado School of Medicine, Aurora, CO, USA, 5Department of Biology, Johns Hopkins University, Baltimore, MD, USA. How the plasma membrane is bent to accommodate clathrin-independent endocytosis is poorly understood. Recent studies suggest the exogenous clathrin independent cargo molecules Shiga toxin and cholera toxin induce the negative membrane curvature required for endocytic uptake by binding and crosslinking multiple copies of their glycosphingolipid receptors on the plasma membrane. But it remains unclear if toxin-induced sphingolipid crosslinking provides sufficient mechanical force for deforming the plasma membrane, or if host cell factors also contribute to this process. To test this, we imaged the uptake of cholera toxin B-subunit into surface-attached tubular invaginations in live cells. We found that a cholera toxin mutant that binds to only one glycosphingolipid receptor accumulates in tubules, and that toxin binding is 353a entirely dispensable for membrane tubulations to form. Unexpectedly, the driving force for tubule extension was found to be supplied by the combination of microtubules, dynein, and dynactin, thus defining a novel mechanism for generation or extension of membrane curvature during endocytic uptake at the plasma membrane. 1773-Plat High-Speed Atomic Force Microscopy of ESCRT Protein Assembly Lorena Redondo1, Nicolas Chiaruttini2, Atsushi Miyagi1, Adai Colom1,2, Aure´lien Roux2, Simon Scheuring1. 1 U1006, INSERM / Aix-Marseille Universite´, Marseille, France, 2 Department of Biochemistry, University of Geneva, Geneva, Switzerland. The endosomal sorting complex required for transport (ESCRT) mediates membrane remodelling in cells. When ESCRT oligomerize, it is able to bud the membrane forming constriction necks that will break resulting in vesicular bodies or the viral envelope, to name a few of its implications. So far, relatively little is known about the molecular fine structure and less about the dynamics of ESCRT assembly, essential for our understanding how it deforms and cleaves the membrane. In this work, we used high-speed atomic force microscopy (HS-AFM) to study the ESCRT machinery, in particular the ESCRT-III complex, Snf7. HS-AFM allows simultaneous observation of structure, dynamics and function of biological assemblies, with nanometer spatial and sub-second temporal resolution. We show HS-AFM movies of the Snf7 complex formation and its dynamics from filament to the maturated circular assembly around the membrane constriction site. We observe interfilament dynamics that provide a basis for a mechanistic explanation how the machinery creates tension for membrane fission by a buckling mechanism. 1774-Plat Mechanisms of Membrane Shaping by Peripheral Proteins Tobias Baumgart. Chemistry, University of Pennsylvania, Philadelphia, PA, USA. Membrane curvature has developed into a forefront of membrane biophysics. Numerous proteins involved in membrane curvature sensing and membrane curvature generation have recently been discovered, and the structure of these proteins and their multimeric complexes is increasingly well-understood. Substantially less understood, however, are thermodynamic and kinetic aspects and the detailed mechanisms of how these proteins interact with membranes in a curvature-dependent manner. New experimental approaches need to be combined with established techniques to be able to fill in these missing details. Here we use model membrane systems in combination with a variety of biophysical techniques to characterize mechanistic aspects of the function of peripheral proteins such as BAR domains, ENTH domains, and synucleins. This includes a characterization of membrane curvature sensing and curvature generation. We also establish kinetic and thermodynamic aspects of BAR protein dimerization in solution, and investigate kinetic aspects of membrane binding. We present two new approaches to investigate membrane shape instabilities leading to stable membrane curvature. We demonstrate that membrane shape instabilities can be controlled by factors such as protein binding, lateral membrane tension, lipid shape and asymmetric bilayer distribution, and macromolecular crowding on the membrane. Our findings are relevant to the mechanistic understanding of membrane trafficking phenomena, including endocytosis. 1775-Plat Role of Hemagglutinin Palmitoylation in Assembly and Fusion of Influenza Virus-Like Particles Petr Chlanda, Elena Mekhedov, Hang Waters, Paul S. Blank, Josh Zimmerberg. NICHD, National Institutes of Health, Bethesda, MD, USA. Influenza A virus is a major human pathogen causing annual epidemics and occasional pandemics. Hemagglutinin (HA), the influenza virus fusion protein, contains on its cytoplasmic tail three conserved cysteins which are palmitoylated. Contradictory data have been reported regarding the role of HA palmitoylation in either membrane fusion or virion assembly. Here we analyzed the role of HA palmitoylation on assembly and fusion of influenza virus-like particles (VLPs) by using fusion assay, cryo-electron microscopy (cryo-EM) and cryo-electron tomography (cryo-ET). VLP assembly, release, morphology, as well as glycoprotein spacing on the surface of the VLP, were not affected by mutation of all three cysteins. However, using both cell-cell and VLP-cell fusion assays we found that palmitoylation plays a role in fusion pore enlargement. We tested HA from three different influenza strains (H2 (A/Japan/305/ 57), H3 (A/Aichi/2/68), H3 (A/Udorn/72)). In all cases HA depalmitoylation impaired pore enlargement, suggesting that the role of palmitoylation in 354a Tuesday, February 10, 2015 membrane fusion is viral strain independent. Cryo-ET of fusion products between liposomes and isolated VLP containing depalmitoylated HA revealed accumulation of small arrested fusion pores with an average internal diameter of 2 nm. 1776-Plat Prefusion Structures of Lipid-Bound SNARE Proteins Suggest Folding Pathways of Trans-SNARE Complex Binyong Liang, Volker Kiessling, Damian Dawidowski, David S. Cafiso, Lukas K. Tamm. University of Virginia, Charlottesville, VA, USA. The assembly of three neuronal soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) proteins synaptobrevin 2, syntaxin-1A, and SNAP-25 is the key step that leads to exocytotic fusion of synaptic vesicles. In the fully assembled SNARE complex, these three proteins form a coiled-coil four-helix bundle structure by interaction of their respective SNARE motifs. Although biochemical and mutational analyses strongly suggest that the heptad-repeat SNARE motifs zipper into the final structure in the N- to C-terminal direction, little is known about the prefusion state of individual membrane-bound SNAREs and whether they change conformation from the unzippered prefusion to the zippered postfusion state in a continuous or step-wise fashion in membrane environments. We have solved the solution NMR structures of micelle-bound synaptobrevin and syntaxin-1A in their prefusion conformations. In addition to their respective transmembrane helices, the SNARE motifs of both proteins have considerable degrees of helical content. For synaptobrevin, only the N-terminal half (residues 36-54) of the SNARE motif forms a transient helix, and the fraction of helical content and interfacial association decreases as the protein is moved from micelle to bicelle to bilayer environments, suggesting that membrane curvature affects the folding of synaptobrevin. For syntaxin, the SNARE motif consists of two well-ordered, membrane-bound helices separated by the ‘‘0-layer’’ residue. These unexpected structural orders of the N- and C-terminal halves of the prefusion SNARE motifs suggest the formation of partially zippered SNARE complex intermediates. Interferometric fluorescence measurements in lipid bilayers confirm that the open SNARE motif helices interact with lipid bilayers and that the assembly of SNARE complexes involves the segmented movements of N- and C-terminal halves of SNARE motifs in relation to the membrane surface. 1777-Plat Energetics and Kinetics of SNARE Zippering and Regulation Revealed by Single-Molecule Manipulation Approach Yongli Zhang. Cell Biology, Yale University, New Haven, CT, USA. Soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins are evolutionarily conserved molecule machines that couple their folding/ assembly to drive diverse intracellular membrane fusion. In particular, neuronal SNAREs mediate extremely fast and calcium-triggered fusion of synaptic vesicles in nerve endings for neurotransmission. It remains unclear how SNAREs fold and how such folding is coupled to membrane fusion and regulated in response to different stimuli. To address these questions, we have characterized the folding energy and kinetics of four representative SNARE complexes at a single-molecule level using high-resolution optical tweezers. Despite their dramatically different fusion rate, all four SNARE complexes assemble by the same step-wise zippering mechanism: slow N-terminal domain (NTD) association, a pause in a force-dependent half-zippered intermediate and fast C-terminal domain (CTD) zippering. However, the energy release from CTD zippering differs from 13 kBT to 28 kBT for yeast and neuron SNARE complexes, respectively. We suggest that SNARE complexes share a conserved zippering pathway to efficiently drive membrane fusion, but release different amount of energy to control the fusion speed. Finally, we will show our latest results about the roles of SNARE mutations, Munc18-1, complexin, and Vc peptides in SNARE zippering and membrane fusion. 1778-Plat Temporally Resolving Protein and Lipid Colocalization at Exocytic Sites in INS-1 Cells Adam J. Trexler, Justin Taraska. National Heart Lung and Blood Institute, National Institutes of Health, Bethesda, MD, USA. The controlled release of material from cells, called exocytosis, is critical for many life processes ranging from synaptic transmission to hormone secretion. Exocytosis is mediated by the assembly of SNARE proteins into a complex that links vesicular and plasma membranes and promotes membrane fusion. Despite a detailed understanding of SNARE complex assembly, a comprehensive description of the arrival and departure of the myriad of other proteins involved in exocytosis is lacking. Here, we describe the localization and dynamics of over a dozen proteins at sites of dense core vesicle exocytosis inside living INS-1 cells. We use two color total internal reflectance fluorescence microscopy to visualize transiently transfected, fluorescently tagged proteins. NPY-GFP, a dense core vesicle cargo protein, is used to localize vesicles and temporally align many exocytic events across several cells. mCherry is used to localize a particular exocytic protein of interest relative to the dense core vesicle and determine its temporal behavior relative to the moment of fusion. Intriguingly, we find that both positive and negative regulators of SNARE complex assembly appear to be present at exocytic sites and diffuse away after fusion occurs. This data suggests that effectors acting on the SNARE complex must be biochemically or conformationally regulated; they are not spatially regulated by exclusion or inclusion at exocytic sites. We are currently examining one exocytic protein, tomosyn, to determine how this protein might be regulated. We are also investigating lipid dynamics at the site of exocytosis using fluorescently tagged lipid binding domains. Our imaging approach should show whether critical lipids are locally synthesized or laterally diffuse to the site of exocytosis. With our data we seek to build a thorough description of protein and lipid behavior at the moment of exocytosis. 1779-Plat Defining a Retrovirus Entry Site by Single Particle Tracking Gregory Melikian1, Sergi Padilla-Parra2, Naoyuki Kondo3, Mariana Marin1. 1 Pediatrics, Emory University, Atlanta, GA, USA, 2The Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, United Kingdom, 3 Institute of Biomedical Science, Kansai Medical University, Osaka, Japan. Current knowledge of viral entry pathways and therefore of host factors involved in virus trafficking to the sites of entry is limited. We imaged single Avian Sarcoma and Leukosis Virus (ASLV) co-trafficking with markers for early (Rab5) and late (Rab7) endosomes and visualized the acidification of endosomal lumen and subsequent virus-endosome fusion. The recruitment of Rab5 marker by virus-carrying endosomes usually coincided with acidification of their lumen, which triggered ASLV fusion. Fusion measured by the viral content release occurred either in early (Rab5-positive) or intermediate (Rab5- and Rab7-positive), maturing endosomes. Expression of different isoforms of the cognate ASLV receptor (TVA) on target cells mediated virus entry from distinct compartments. In cells expressing the transmembrane TVA950 receptor, ASLV preferentially entered and fused with slowly maturing early endosomes, which accumulated Rab7 after a considerable delay. Viral entry occurred from either slowly or quickly maturing endosomes in cells expressing the GPI-anchored TVA800 receptor, as manifested by ASLV fusion with early (Rab5-positive) or intermediate (Rab5- and Rab7-positive) compartments. Simultaneous visualization of endosome acidification and viral content release enabled the measurement of the true kinetics of viral fusion with endosomes, which was independent of the receptor isoform. We concluded that the sites of ASLV entry are determined by the kinetic competition between endosome maturation and low pH-dependent fusion. These findings demonstrate the ability of ASLV to enter cells via alternative endocytic pathways and establish infection by fusing with distinct endosomal compartments. This work was partially supported by the NIH R01 AI053668 grant. Tuesday, February 10, 2015 Platform: Force Spectroscopy and Scanning Probe Microscopy 1780-Plat Imaging and Three-Dimensional Reconstruction of Chemical Groups in a Protein Complex using DNA Labels Duckhoe Kim1, Ozgur Sahin1,2. 1 Biological Sciences, Columbia University, New York, NY, USA, 2Physics, Columbia University, New York, NY, USA. Atomic force microscopy (AFM) is a powerful tool for imaging and chemical characterization of bio-samples at molecular resolution in physiologicallyrelevant environments. However, the localized tip-sample interactions limit high-resolution images to the topmost layer of surfaces. Consequently, characterizing the three-dimensional (3-D) inner structures of molecules has been a challenge. Here, we demonstrate three-dimensional (3-D) localization of chemical groups within a single protein complex using AFM. We employ short DNA sequences to label specific chemical groups inside a protein complex. T-shaped cantilevers functionalized with complementary probe DNAs allow locating each label with sequence specificity and sub-nanometer resolution. We also measure pairwise distances between labels and reconstruct the 3-D loci of the target groups using simple geometric calculations. Experiments with the biotin-streptavidin complex showed that the 3-D loci of carboxylic acids of biotins are within 2-Angstroms of their respective 3-D loci in the corresponding crystal structure, suggesting AFM may complement existing structural biological techniques in solving structures that are difficult to study due to their size and complexity. This technique maybe finds applications in studying structure of DNA or RNA binding proteins. 1781-Plat Acoustic Force Spectroscopy Douwe Kamsma1, Gerrit Sitters1, Gregor Thalhammer2, Monika Ritsch-Marte2, Erwin J.G. Peterman1, Gijs J.L. Wuite1. 1 Physics and astronomy, VU University Amsterdam, Amsterdam, Netherlands, 2Department fu¨r Physiologie und Medizinische Physik, Medizinische Universita¨t Innsbruck, Innsbruck, Austria. Single-molecule force spectroscopy has become an indispensable tool to unravel the structural and mechano-chemical properties of biomolecules. In most forcespectroscopy instruments only a limited number of biomolecules can be studied simultaneously, which reduces experimental throughput and limits statistics. At the same time, many independent measurements are required to distinguish the intrinsic stochasticity of the process of interest from heterogeneity. With Acoustic Force Spectroscopy (AFS) we extend the force-spectroscopy toolbox with an acoustic manipulation device that allows exerting acoustic forces on tethered molecules. AFS is a Lab-on-a-chip device consisting of a flow cell of two glass plates with a fluid chamber in between and a piezo element glued on top. While applying an alternating voltage to the piezo element, forces from sub-pN to hundreds of pNs are exerted to thousands of biomolecules in parallel, with sub-millisecond response time and inherent stability. As a proof of concept we performed force-extension measurements on DNA and RecA-coated DNA. These experiments demonstrate that AFS can be used to apply highly controlled forces up to at least 120 pN, with a force ramp speed between 104 - 102 pN/s and showing inherent stability over tens of hours over an observation area of at least 1 mm2. AFS experiments are highly parallel, allowing the simultaneous measurement of thousands of biomolecules simultaneously, in a single field of view. We demonstrate the use of this by mapping the energy landscape of the DIG/anti-DIG antibody-antigen bond over 6 orders of magnitude of force loading rates within 2 days of experimentation. AFS distinguishes itself by its relative simplicity, low cost and compactness, which allow straightforward implementation in lab-on-a-chip devices. These aspects will help to spread single-molecule methods from the realm of fundamental research in specialized laboratories towards more wide-spread applications in for example molecular biology and medical diagnostics. 1782-Plat Revisiting the Free Energy of Modular Proteins under Force Ionel Popa1, Jaime Andre´s Rivas-Pardo1, Edward C. Eckels1, Jessica Valle-Orero1, Thomas B. Kahn1, Ronen Berkovich2, Guillaume Stirnemann3, Hu Chen4, Vicente I. Fernandez5, Bruce J. Berne3, Jie Yan6, Julio M. Fernandez1. 1 Biological Sciences, Columbia University, New York, NY, USA, 2Chemical Engineering, Ben-Gurion University of the Negev, Beer-Sheva, Israel, 3 Chemistry, Columbia University, New York, NY, USA, 4Physics, Xiamen University, Xiamen, China, 5Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA, 6Physics, National University of Singapore, New York, Singapore. 355a The elasticity of muscle tissue relies on tandem modular proteins such as titin, which gives muscles passive elasticity. A free-energy model is the only way to understand and predict the behavior under force of such complex proteins, composed of hundreds of individual domains. Here we use magnetic tweezers to measure the dynamics of single tandem modular proteins under constant force conditions during hour-long recordings. At forces between 4 to 17 pN we measure unfolding/refolding of individual domains as upward/downward steps in the end-to-end protein length, while higher forces yield unfolding steps exclusively. We find a strong force dependency of the step size of proteins undergoing folding/unfolding reactions with the applied force. This finding contradicts current free energy models of proteins that typically do not consider the polymeric nature of a denatured polypeptide chain under force and simply scale the free energy of a protein with the mechanical work. To explain the measured step size dependency with force we propose a new free energy model that also considers the entropic work needed to extend the molecule. Brownian dynamics simulations over the proposed free energy landscape accurately reproduce our experimental benchmarks. The experimental and theoretical advances demonstrated in this work provide a novel view on the free energy of proteins under force, now permitting a more realistic modeling of tissue elasticity. 1783-Plat Directly Observing the Reversible Unfolding and Refolding of an Alpha/ Beta Protein by Single-Molecule Atomic Force Microscopy Chengzhi He1, Chunguang Hu2, Xiaodong Hu2, Xiaotang Hu2, Adam Xiao1, Hongbin Li1,2. 1 Chemistry, The University of British Columbia, Vancouver, BC, Canada, 2 State Key Laboratory of Precision Measurements Technology and Instruments, Tianjin University, Tianjin, China. Single-molecule force spectroscopy based on atomic force microscopy (AFM) has evolved into a powerful tool to study protein folding-unfolding dynamics and mechanism. Direct observation of protein refolding in real time using AFM has been challenging. Proteins that have been observed to refold using AFM are mainly limited to all-alpha proteins, such as ankyrin and calmodulin. Here we report the use of AFM to directly monitor the folding of an alpha/beta protein, NuG2. Our results indicate that at slow pulling speeds (<50 nm/s), the refolding of NuG2 can be clearly observed. Lowering the pulling speed reduces the difference between the unfolding forces and refolding forces, bringing the non-equilibrium unfolding-refolding towards equilibrium. At very slow pulling speeds (~2 nm/s), reversible unfolding and refolding were observed. Based on Crooks Theorem, we measured the equilibrium free energy change between the folded and unfolded states of NuG2, which is in good agreement with values reported using bulk chemical denaturation method. Our results demonstrate the utility of AFM in elucidating the unfolding-refolding dynamics of proteins close to equilibrium. 1784-Plat Electromagnetic Tweezers with Independent Force and Torque Control Chang Jiang, Troy A. Lionberger, Diane M. Wiener, Edgar Meyho¨fer. University of Michigan, Ann Arbor, MI, USA. Mechanical forces and torques play an important role in many biological processes. A range of instruments have been developed for direct and precise measurements of force and torque applied by biomolecules ranging from proteins to nucleic acids. However, simultaneous force and torque applications in which the two components are decoupled and controlled independently remain challenging and have only been implemented by recently-developed hybrid magnetic tweezers combining electro- and permanent magnets. Our goal was to develop bona fide electromagnetic tweezers (eMT) that can apply force and torque on single biomolecules or polymer molecules conjugated via superparamagnetic microspheres and can control the two components independently simply by changing the currents applied to different coils of the eMT. We implemented our eMT by combining a monopole that generates a force and a set of quadrupoles that generate a torque. To demonstrate the capability of our tweezers we attached Janus beads to single DNA molecules. We show that tension in the piconewton force range can be applied to single DNA molecules and simultaneously the molecule can be twisted with torques in the piconewtonnanometer regime. Our results also demonstrate that the two components are independently controlled. At various force levels applied to the Janus bead, the trap torsional stiffness can be changed simply by varying the current magnitude applied to the quadrupole torque tweezers. Our eMT voids the need of complex positioners to translate and rotate permanent magnets or frequency modulation of current to decouple the force and torque used in previous works. We believe that the flexible control of the mechanics of biomolecules with our eMT will enable novel studies of DNA-protein interactions and DNA conformation dynamics. 356a Tuesday, February 10, 2015 1785-Plat Surface-Free Single-Molecule Force Spectroscopy Yan Jiang1,2, Wesley Wong1,2. 1 PCMM, Boston Children’s Hospital, Boston, MA, USA, 2Bcmp, Harvard Medical School, Boston, MA, USA. Mechanical forces play key roles throughout biology. Most existing methods for probing forces in/between molecules and cells, such as the AFM, optical tweezers, magnetic tweezers, traction force microscopy, etc., require molecules to be tethered to the surfaces of cover glasses or beads. However, surface tethering has a number of drawbacks. First, the conjugation chemistry for tethering can be challenging. Second, the spatial confinement and the nonspecific attraction from the surface can induce artifacts to the dynamics of the molecule. For semi-rigid objects, such as actin filaments, surface attachment could also induce bending and twisting of the filament, complicating the applied force profile. To overcome these issues, we present a surface-free force spectroscopy method that uses a high-speed cross-slot hydrodynamic trap, capable of stretching molecules and cells with hydrodynamic drag. The trap is based on a glass microfluidic cross-slot flow chamber. Buffer flows in from two opposite directions and exits via the two orthogonal outlets to create an elongational flow field with a stagnation point in the center. As a result, objects near the stagnation point are stretched by the viscous drag from the flow. In addition, the pressure in one of the outlet reservoirs is electronically controlled with a high-speed feedback algorithm to stabilize the object at the stagnation point. Thanks to the high-speed feedback, we can apply much higher flow rate and therefore much higher stretching force on the trapped object than with the cross slot alone. We demonstrate tension-dependent actin severing, extension of von Willebrand Factor (a key protein in haemostasis that changes conformation in response to hydrodynamic stress in blood stream), overstretching of double strand DNA, and stretching deformation of red blood cells. In summary, the high-speed cross-slot hydrodynamic trap can be a powerful, surface-free alternative to more commonly used force spectroscopy methods. 1786-Plat High-Speed Force Spectroscopy Unbinds Streptavidin-Biotin at the Velocity of Molecular Dynamics Simulations Felix Rico1, Andreas Russek2, Helmut Grubmueller2, Simon Scheuring1. 1 U1006 Inserm & Aix-Marseille Universite´, Marseille, France, 2Theoretical and Computational Biophysics, Max Planck Institute for Biophysical Chemistry, Goettingen, Germany. The forced disruption of the (strept)avidin-biotin complex by atomic force microscopy (AFM) and other techniques opened the field and established the basis of single molecule force spectroscopy (1-3). Steered molecular dynamics (SMD) simulations provided atomic description of the unbinding process (4). However, the maximum experimental AFM velocities were typically of 10 mm/s, while SMD simulations were performed in the range of 1 m/s to 10 m/s. Recent development of high-speed force spectroscopy (HS-FS) using HS-AFM allowed velocities in the mm/s range (5). We have applied HS-FS to probe the binding strength of the streptavidin-biotin complex at velocities up to ~8 mm/s, paralleled by SMD simulations. The experimental dynamic force spectrum of the unbinding process is compared with SMD simulations. The combination of HS-FS and SMD at overlapping velocities provides an atomic description of the unbinding process measured by experiment. 1. Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature 397:50-53. 2. Florin, E. L., V. T. Moy, and H. E. Gaub. 1994. Adhesion Forces Between Individual Ligand-Receptor Pairs. Science 264:415-417. 3. Lee, G. U., D. A. Kidwell, and R. J. Colton. 1994. Sensing Discrete Streptavidin Biotin Interactions with Atomic-Force Microscopy. Langmuir 10:354-357. 4. Grubmuller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: Molecular mechanics calculation of the streptavidin biotin rupture force. Science 271:997-999. 5. Rico, F., L. Gonzalez, I. Casuso, M. Puig-Vidal, and S. Scheuring. 2013. High-Speed Force Spectroscopy Unfolds Titin at the Velocity of Molecular Dynamics Simulations. Science 342:741-743. 1787-Plat The Princess and the Pea: A Story of Cell Mechanics Mehdi Roeinpeikar, Qian Xu, Xuefeng Wang, Taekjip Ha. University of Illinois at Urbana-Champaign, Urbana, IL, USA. Single molecules of integrins, a class of trans-membrane proteins involved in cell adhesion, experience a peak force of 40 pN through their ligands during initial adhesion. This force requirement was determined using a series of double stranded (ds) DNA tethers, each with a different rupture force, conjugated to the ligand. Here, force spectroscopy is performed by multiplexing two types of DNA tethers on the same surface: one that requires a strong rupture force (54 pN) and the other, a weak rupture force (12 pN). When presented alone, cells adhere to the strong tethers but not the weak tethers. However, when multiplexed, the result is synergistic; cells can adhere to a surface displaying just a few molecules of the strong tether if, and only if, they are presented along with many weak tethers. This degree of ultra-sensitivity raises a question on how cells suppress noise during decision making based on nano-mechanical environments. Platform: Protein-Small Molecule Interactions 1788-Plat Biasing Potential Replica Exchange Multi-Site l-Dynamics for Efficient Free Energy Calculations of Protein-Ligand Interactions Kira A. Armacost, Garrett B. Goh, Charles L. Brooks. Department of Chemistry, University of Michigan, Ann Arbor, MI, USA. Traditional free energy calculations are known for their issues with scalability, speed and convergence. We have recently developed the biasing potential replica exchange multi-site l-dynamics (BP-REX MSlD) method, which is a free energy method capable of transforming multiple substituents at multiple sites on a common framework with relative ease. With this methodology, we are able to show convergence of flexible moieties for symmetric benzoquinone derivatives, and have developed a series of metrics capable of increasing the l-space sampling. We grouped and ordered the substituents based on volume occupancy, and were able to compute the free energy of binding for a series of challenging geldanamycin-derivatives for heat shock protein 90. The perturbations spanned ˚ 3 and we were able to model these with a 2.4 kcal/mol average by as much as 60 A unsigned error. These metrics coupled with BP-REX MSlD allow for routine calculations on the order of hundreds of compounds in a few simulations. 1789-Plat Survey of Phosphorylation Near Drug Binding Sites in the Protein Data Bank (PDB) and their Effects Kyle P. Smith, Kathleen M. Gifford, Joshua S. Waitzman, Sarah E. Rice. Cell & Molecular Biology, Northwestern University, Chicago, IL, USA. While it is currently estimated that 40-50% of eukaryotic proteins are phosphorylated, little is known about the frequency and local effects of phosphorylation near pharmaceutical inhibitor binding sites. In this study, we investigated how frequently phosphorylation may affect the binding of drug inhibitors to target proteins. We examined the 453 non-redundant structures of soluble mammalian drug target proteins bound to inhibitors currently available in the Protein Data Bank (PDB). We cross-referenced these structures with phosphorylation data available from the PhosphoSitePlus database. 322/453 (71%) of drug targets have evidence of phosphorylation that has been validated by multiple methods or labs. For 132/453 (29%) of those, the phosphorylation site is ˚ of the small molecule-binding site, where it would likely alter small within 12A molecule binding affinity. We propose a framework for distinguishing between drug-phosphorylation site interactions that are likely to alter the efficacy of drugs vs. those that are not. In addition we highlight examples of wellestablished drug targets, such as estrogen receptor alpha, for which phosphorylation may affect drug affinity and clinical efficacy. Our data suggest that phosphorylation may affect drug binding and efficacy for a significant fraction of drug target proteins. 1790-Plat Identification and Characterization of Protein-Protein Interaction Effectors Targeting the Invasion Machinery of the Malaria Parasite Lauren E. Boucher1,2, Christine S. Hopp2,3, Photini Sinnis2,3, Ju¨rgen Bosch1,2. 1 Department of Biochemistry and Molecular Biology, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA, 2Johns Hopkins Malaria Research Institute, Baltimore, MD, USA, 3Department of Microbiology and Molecular Immunology, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA. Emerging resistance of Plasmodium falciparum to current treatments is concerning and requires the development of a new generation of drugs. In pursuit of novel compounds, we have targeted protein-protein interactions (PPIs) essential to the parasite. Plasmodium uses an actomyosin motor, part of the glideosome complex, for gliding and invasion of host cells. A key interaction of the glideosome is between aldolase and the cytoplasmic tail of the thrombospondin-related adhesive protein, TRAP. It is thought that the dynamic aldolase-TRAP complex must dissociate for forward motility and invasion to progress; therefore, we have developed an assay to identify compounds that promote this interaction, resulting in stalled gliding and invasion. Tuesday, February 10, 2015 We produced MBP-tagged and biotinylated peptides for studying the interaction between aldolase and TRAP using surface plasmon resonance. We have screened the Medicines for Malaria Ventures library and have identified several compounds with concentration-dependent effects in vitro. Five promising compounds were moved forward into parasite gliding and invasion assays. We have observed phenotypic differences in the ability of Plasmodium parasites to glide, and found a reduced gliding velocity and abnormal rates of sporozoite attachment and detachment. To determine the effect on invasion, both red blood cell and hepatocyte invasion assays have been performed. We are currently attempting to determine a co-crystal structure of the ternary aldolase-TRAP-compound complex, and to move promising compounds into an in vivo mouse model. 1791-Plat Absolute Binding Free Energy Calculations of Bromodomain Inhibitors Matteo Aldeghi1, Stefan Knapp2, Alexander Heifetz3, John J. Barker3, Michael J. Bodkin3, Richard J. Law3, Philip C. Biggin1. 1 Structural Bioinformatics and Computational Biochemistry, Department of Biochemistry, University of Oxford, Oxford, United Kingdom, 2Structural Genomics Consortium and Target Discovery Institute, University of Oxford, Oxford, United Kingdom, 3Evotec (U.K.) Ltd., Abingdon, United Kingdom. Computational approaches have been increasingly employed for drug discovery in both academia and industry. The ultimate goal of rational drug design is the prediction of the biological activity of a compound, and such activity is driven, at the molecular level, by the specific intermolecular interactions between the small organic molecule, the biological target and the solvent. Similar molecular recognition processes are crucial for many biological functions and the strength of the recognition is characterized by its binding affinity. Thanks to important advances in theory and computing in the last decades, predictions of binding affinities using physics-based simulations are gaining popularity. In particular, binding free energy estimates based on alchemical pathways have been shown to be a rigorous approach for the affinity prediction problem and hold the promise to be able to guide lead optimisation. However, whilst relative calculations have made significant contributions in a drug-discovery context, absolute calculations have so far been mostly applied to model systems despite the advantages related to the ability to estimate affinities for largely diverse sets of molecules. We present the results of a study where we evaluated the performance of alchemical free energy estimates for a set of drug-like molecules that have been developed to target bromodomains, a family of epigenetic marks readers with established therapeutic potential. The study evaluates the performance of the protocol employed from a retrospective and perspective standpoint, with particular attention to the precision of the calculations and comparing the theoretical results with high-quality binding data. We show that, for this epigenetic target, good agreement between calculations and experiments is achievable even for very challenging compounds, suggesting that alchemical free energy calculations might be approaching the degree of reliability required in order to have an impact in drug discovery campaigns. 1792-Plat Positive Modulators of Glycine Receptors with Analgesic Potential Identified by Virtual Screening Marta M. Wells, David D. Mowrey, Edom Seyoum, Tianmo Sun, Yan Xu, Pei Tang. Anesthesiology, University of Pittsburgh, Pittsburgh, PA, USA. Human glycine receptors (hGlyRs) are important targets for neuroactive drugs, including for analgesic therapies. A previous study has identified the crucial role of residue S296 in hGlyR-a1 potentiation by D9-tetrahydrocannabinol (THC) and in cannabinoid-induced analgesia. The recently determined NMR structure of the hGlyR-a1 transmembrane domain (TMD) provides a basis for structure-based virtual screening to discover new analgesic drugs. Using a large ensemble of hGlyR-a1 structures generated from molecular dynamics simulations based on the NMR structure, we screened 1549 FDA approved compounds from the DrugBank database targeted to a cannabinoid binding site near residue S296. Drugs were ranked based on their predicted binding affinities across the ensemble of hGlyR-a1 TMD structures. Four leading compounds were selected for experimental validation in Xenopus laevis oocytes expressing hGlyR-a1. At a low concentration (1 mM), all four leading compounds potentiate hGlyR-a1 currents more than two times, which was greater than that of THC. The hit rate is remarkable. The study provides strong evidence that these leading compounds will be at least as effective as THC for analgesia by acting on hGlyR-a1, but without psychoactive effects. The protocol developed here can easily be applied to the discovery of novel analgesic compounds of even higher efficacy on hGlyRs. Research supported by grants from the NIH and XSEDE. 357a 1793-Plat Green and Black Tea Polyphenols Mechanistically Inhibit the Aggregation of Amyloid-b in Alzheimer’s Disease Shelby E. Chastain1, Melissa Moss2. 1 Biomedical Engineering, University of South Carolina, Columbia, SC, USA, 2 Chemical Engineering, University of South Carolina, Columbia, SC, USA. Alzheimer’s disease (AD) is the 6th leading cause of death and is the only disease among the top 10 that cannot be prevented, cured or treated. The amyloid cascade hypothesis states that naturally occurring amyloid-b (Ab) monomer aggregates via a nucleation-dependent pathway to form soluble aggregates and insoluble fibrils that deposit as plaques in the brain. Consequently, inhibition of Ab aggregation is one therapeutic strategy for AD. This study sought to explain epidemiological correlations between frequent tea consumption and reduced incidence of AD by identifying the Ab aggregation inhibitory capabilities of key polyphenol components in green and black tea. Polyphenols studied include green tea catechins, epicatechin, epigallocatechin and epigallocatechin gallate, as well as black tea theaflavins, theaflavin and theaflavin monogallate. Four assays were used to target unique steps along the aggregation pathway: a monomer aggregation assay to monitor the overall aggregation process; an oligomerization assay to monitor the initial nucleation step; an association assay to monitor the late stage lateral binding of soluble aggregates; and an elongation assay to monitor the late stage lengthening of soluble aggregates. Catechins and theaflavins show different inhibitory capabilities at varying mechanistic steps of the Ab aggregation pathway. Catechins affect only the later stages of aggregation, suggesting that catechins may bind a specific structure present in aggregates. Conversely, theaflavins show inhibitory capabilities at every stage of aggregation, alluding to a sequence specific recognition. Furthermore, better inhibitory capabilities, for both polyphenol categories, can be correlated with the number of gallate groups. 1794-Plat Effect of Reactive Aldehydes on Ionophore-Mediated Transmembrane Translocations of HD and KD Alina A. Pashkovskaya, Elena E. Pohl. Univ Vet Medicine, Vienna, Austria. Reactive aldehydes (RA) are involved in the onset and progression of many pathologies such as cardiovascular and neurodegenerative diseases. The precise mechanisms by which aldehydes contribute to these diseases remain unclear. Previously, we and other groups suggested that RAs modify the membrane protein function by either binding to the proteins directly(1) or to aminophosphosholipids, with a subsequent alteration in protein conformation. Here, we investigated the influence of biologically important RAs such as 4-hydroxy-2-nonenal (HNE), 4-oxo-2-nonenal (ONE) and 4-hydroxy-2-hexenal (HHE) on the transport kinetics of the ionophores carbonyl-cyanide-mchlorophenylhydrazone (CCCP) and valinomycin (Val). We found that RA increases the membrane conductance, G, in the presence of Val in the following order ONE>HNE>HHE. In contrast G decreases in the presence of CCCP and ONE. The presence of phosphatidylethanolamine in the membrane was crucial for the effect to occur. The results are consistent with the hypothesis that RA adducts alter membrane boundary potential. 1. E. A. Malingriaux, et al., ‘‘Fatty Acids are Key in 4-Hydroxy-2-NonenalMediated Activation of Uncoupling Proteins 1 and 2’’ PLoS. ONE. 8(10), e77786 (2013). 1795-Plat Developing High-Throughput Fluorescence-Based Assays for Measuring Kinase Inhibitor Free Energies of Binding Sonya M. Hanson, Jan-Hendrik Prinz, Julie M. Behr, Patrick B. Grinaway, Arien S. Rustenburg, Kyle A. Beauchamp, Daniel L. Parton, John D. Chodera. Computational Biology, Memorial Sloan Kettering Cancer Center, New York, NY, USA. Understanding the specificity of kinase inhibitors has tremendous therapeutic significance. To predict inhibitor selectivity computationally, an iterative approach incorporating experimental measurements is ideal. This work describes the development of a high-throughput label-free fluorescent ligandbinding assay to measure inhibitor affinities. Taking advantage of the intrinsic fluorescence increase of a group of FDA-approved kinase inhibitors upon binding kinases, we are able to measure inhibitor binding affinity with small amounts of protein and without any fluorescent labels. This facilitates rapid characterization of a wide range of kinase inhibitors and kinase resistance mutants, within a system that can be reproduced identically in molecular dynamics simulations. 358a Tuesday, February 10, 2015 Symposium: Nanoclustering of Membranes and Membrane Proteins 1796-Symp Structure and Function of Membrane-Remodeling ESCRT-III Assemblies John McCullough1, Marissa Saunders1, Leremy Colf1, Wes Sundquist1, Adam Frost2. 1 Biochemistry, University of Utah, Salt Lake City, UT, USA, 2Bioscience, University of Utah, Salt Lake City, UT, USA. The ESCRT pathway mediates a series of important cellular membrane remodeling and fission events, including cytokinetic abscission. During these processes, ESCRT-III family proteins, including CHMP1B and IST1, form filaments that appear to constrict membranes and facilitate fission. Here, we report the first structure of ‘‘open’’ and assembled ESCRT-III proteins. Nearatomic resolution electron cryomicroscopy reveals that filaments comprise a copolymeric assembly of an open conformation inner strand and, unexpectedly, a closed conformation outer strand. 1797-Symp In Vivo-Studies of GPCR Conformational Changes using FluorescenceBased Assays Martin Lohse. Institute of Pharmacology and Toxicology and Rudolf Virchow Center, University of Wurzburg, Wurzburg, Germany. Activation of and signaling by GPCRs involves conformational changes in the receptors themselves as well as downstream signaling proteins and also the rearrangement of protein complexes. We have developed a number of optical assays based on fluorescence resonance energy transfer (FRET) which allow the measurement and imaging of GPCR activation and signaling in intact cells. These assays are based on fluorescently labeled receptors and/or downstream signaling proteins and can be used in intact cells as well as in entire organisms such as Drosophila larvae. They permit the analysis of GPCR activation mechanisms, their amplitudes and kinetics, as well as an analysis of the spatiotemporal patterning of receptor activation and signaling. We have also used single particle tracking to elucidate the movements of these proteins on the cell surface and the monitoring of their interactions. Our data show that these processes are highly dynamic and that they can occur in temporally and spatially restricted patterns. This dynamic behavior begins with the high mobility of the components in the cell membrane and their dynamic and reversible interaction and ends with temporally and spatially regulated activation and signaling. Supported by grants from the Deutsche Forschungsgemeinschaft (SFB487 and SFB688), the European Research Council (Grants ‘‘Topas’’ and Fresco), the Humboldt Foundation and the European Molecular Biology Organization (EMBO). 1798-Symp Lipid Organization of the Plasma Membrane Helgi I. Ingolfsson1, Peter Tieleman2, Siewert Marrink1. 1 Biophysical Chemistry, Univ Groningen, Groningen, Netherlands, 2Centre for Molecular Simulation, Univ Calgary, Calgary, Netherlands. The detailed organization of cellular membranes remains rather elusive. In this lecture, I provide a high-resolution view of the lipid organization of a plasma membrane (PM) that is based on large-scale molecular dynamics simulations using the Martini force field [1]. Our mammalian PM model consists of 63 different lipid species, combining 14 types of headgroups and 11 types of tails asymmetrically distributed across the two leaflets [2]. The complexity of the PM mixture gives rise to an enrichment of cholesterol in the outer leaflet and a general non-ideal lateral mixing of the different lipid species. Transient domains with liquid-ordered character form and disappear on the microsecond time scale. These domains are coupled across the two membrane leaflets. In the outer leaflet, distinct nanodomains consisting of gangliosides are observed. Additional simulations of membrane proteins embedded in the PM reveal preferential interactions with specific lipids. The in-silico data provide a key view on the lateral organization of lipids and proteins in one of life’s fundamental structures, the cell membrane. [1] S.J. Marrink, D.P. Tieleman. Perspective on the Martini model. Chem. Soc. Rev., 42:6801-6822, 2013. [2] H.I. Ingo´lfsson, M.N. Melo, F.J. van Eerden, C. Arnarez, C.A. Lo´pez, T.A. Wassenaar, X. Periole, A.H. De Vries, D.P. Tieleman, S.J. Marrink. Lipid organization of the plasma membrane. JACS, 2014. http://dx.doi.org/10.1021/ ja507832e 1799-Symp Differential Phosphatidylserine Recognition by the Tim Family of Immune Regulatory Receptors Ka Yee C. Lee. Department of Chemistry, The University of Chicago, Chicago, IL, USA. Recognition of phosphatidylserine (PS) lipids exposed on the extracellular leaflet of plasma membranes is implicated in both apoptotic cell removal and immune regulation. Using a combination of interfacial x-ray scattering, molecular dynamics simulations, and membrane binding assays, we examined how different members of the T-cell immunoglobulin and mucin-domaincontaining (Tim) family of PS receptors recognize PS in the context of a lipid bilayer. Our findings demonstrate that in addition to the known Ca2þ-coordinated, single-PS binding pocket, the different Tim proteins have different number of weaker sites of potential ionic interactions with PS lipids, and show different levels of hydrophobic insertion. These subtle differences in PS recognition likely contribute to the differences in immunological function among the Tim proteins. Symposium: Extremophiles: Testing the Physical Limits of Living Systems 1800-Symp Protein Folding at Extreme Temperatures: Current Issues Georges Feller. Biochemistry, University of Liege, Liege - Sart Tilman, Belgium. The range of temperatures compatible with life is currently estimated from 20 C, as exemplified by metabolically active bacteria between sea ice crystals, and up to 122 C in hydrothermal vents as exemplified by the archaeon Methanopyrus kandleri. Microbial life under these extreme environmental temperatures obviously requires a vast array of adaptations at all cellular levels. In the context of protein folding, as soon as a polypeptide emerges from the ribosome, it is exposed to the effects of the environmental temperatures. Recent investigations have addressed some essential questions: i) what is the effect of extreme environmental temperatures on the protein folding rate; ii) how do PPIases catalyze prolyl isomerization, a rate-limiting step in protein folding; iii) the ‘‘trigger factor’’ is the first chaperone interacting with nascent chains: how does it help protein folding at extreme temperatures and iv) what are the properties of the final native state of proteins adapted to these temperatures? The available results that will be summarized here open new perspectives for the study of life in extreme environments. 1801-Symp Using Single Molecule Force Spectroscopy to Probe Proteins from Extremophiles Lorna Dougan. University of Leeds, Leeds, United Kingdom. Extremophiles are organisms which survive and thrive in the most extreme chemical and physical conditions on Earth. The proteins from extremophilic organisms play a key role in enabling them to survive and function in specific environmental extremes. These proteins are of great interest as they have the ability to retain their folded structure and to possess the necessary flexibility to be functional under conditions which normally denature proteins. For this reason, they offer attractive, model systems in which to explore the origin of protein structure and dynamics under different, extreme environmental conditions. We use single molecule force spectroscopy to measure the mechanical stability and flexibility of proteins derived from extremophile organisms. We have characterised the mechanical stability of a cold shock protein from the hypethermophilic organism, Thermotoga maritima in the temperature range 5-40 C. We measure temperature-dependent changes in features of the unfolding energy landscape of this protein by studying the pulling speed dependence of the unfolding force with temperature in combination with Monte Carlo simulations. We find that the position of the transition state to unfolding shifts away from the native state with increased temperature, reflecting a reduction in the spring constant of the protein and an increase in the malleability of the structure. The mechanical robustness and malleability of this cold shock protein over the temperature range studied, provides an insight into the dynamical properties of hyperthermophilic proteins. To gain further insight into the kinetic stability and adaptation strategies of this protein we are examining structural homologues from mesophilic organisms and mutants of the cold shock protein. These will provide a deeper understanding of the adaptations found in hyperthermophilic proteins, and will enable the rational design of proteins for bionanotechnological applications. Tuesday, February 10, 2015 1802-Symp Mechanisms of Pressure Effects in Biology: From Proteins to Live Bacteria Catherine Ann Royer. Biological Sciences, Rensselaer Polytechnic Institute, Troy, NY, USA. Nearly 80% of the terrestrial biosphere is found in the ocean at depths over 1000 meters. Most of this space is dark and cold, such that the organisms that live there have had to adapt to high pressure, low temperature and lack of light. These parameters are known to have an enormous effect on bio-molecular structure and dynamics, and presumably cellular physiology. In particular, pressures of up to 1000 bar are reached in the deepest parts of the ocean. Understanding how organisms adapt to such extreme environments requires and understanding of how pressure effects bio-molecules and the bio-molecular basis of physiological responses to pressure. We have pursued these questions using model systems and state of the art biophysical approaches. In this talk I will present our recent results on defining the molecular mechanisms of pressure-induced unfolding of proteins and the in vivo response of E. coli to pressure shock. 1803-Symp What Limits Microbial Growth at High Pressure? Doug Bartlett. Scripps Institution of Oceanography, University of California, San Diego, La Jolla, CA, USA. Elevated hydrostatic pressure is an important but relatively unstudied thermodynamic parameter that has influenced the evolution and distribution of life on and in Earth. Piezophiles are deep-sea and deep-subsurface microorganisms whose pressure optima for growth exceeds atmospheric pressure. In this presentation multiple lines of inquiry will be described which provide new details on piezophiles and piezophily. This will include the isolation and characterization of piezophilic and piezotolerant microbes from the Challenger Deep within the Mariana Trench and from the Mid-Cayman Rise hydrothermal vent system. Microbes recently isolated which are capable of growth above the known upper pressure limit for life (130 megapascals) will be described. Comparative analyses of genomes obtained from single cells extracted from trenches, from pure cultures of piezophiles, and from one deep trench metagenome indicate that life at high pressure includes an expansion of regulatory features and of pathways of carbon and energy acquisition. Curiously deep ocean microbes also contain a high proportion of genes encoding aquaporins, channels used for water and solute transport in and out of the cells. The possible significance of this discovery to the interplay between osmotic pressure and hydrostatic pressure will be discussed. Finally, laboratory-based directed evolution experiments will be described in which cells of the mesophile Escherichia coli which are capable of enhanced growth at high pressure and decreased growth at atmospheric pressure have been isolated. Analyses of a mutant derived from this selection indicates that changes in membrane physical state brought about by the introduction of increased proportions of unsaturated fatty acids are necessary but not sufficient for the resulting evolutionary changes driving the cells in the direction of piezophily. The time is now ripe to utilize the tools of biophysics to examine the basis and limits of life at high pressure. Platform: Optical Microscopy and Super-Resolution Imaging II 1804-Plat Background-Free Super-Resolution Microscopy of Subcellular Structures by Lifetime Tuning and Photons Separation Luca Lanzano1, Ivan Coto Hernandez1, Marco Castello1, Enrico Gratton2, Alberto Diaspro1, Giuseppe Vicidomini1. 1 Nanophysics, Istituto Italiano di Tecnologia, Genoa, Italy, 2Laboratory for Fluorescence Dynamics, Department of Biomedical Engineering, University of California, Irvine, CA, USA. The visualization at the nanoscale level inside cells is a fundamental need in molecular biology. The challenge of increasing the spatial resolution of an optical microscope beyond the diffraction limit can be reduced to a spectroscopy task by proper manipulation of the molecular states. The nanoscale spatial distribution of the molecules inside the detection volume of the microscope can be encoded within the fluorescence dynamics and can be decoded by resolving the signal into its dynamics components [1]. We present here a robust and general method, based on the phasor analysis [2], to spatially sort the fluorescent photons on the basis of the associated molecular dynamics and without making use of any fitting procedure. In a specific implementation of this method, we generate spatially controlled gradients in the fluorescence lifetime by stimulated emission [3]. The separation of the time-resolved fluorescence components sorts photons according to their spatial positions. Spatial resolution can be increased indefinitely by increasing the number of resolved components up to a maximum, pre- 359a dictable number, determined by the amount of noise. The method also isolates any uncorrelated background signal. We demonstrate that this spectroscopybased method provides background-free nanoscale imaging of subcellular structures, opening new routes in super-resolution microscopy based on the encoding of spatial information through manipulation of molecular dynamics. We discuss advantages and limitations considering application of the method to the imaging of sparse cytoskeletal structures and large scale organization of chromatin. References [1] Enderlein J. Breaking the diffraction limit with dynamic saturation optical microscopy. Applied Physics Letters 87,094105(2005). [2] Digman MA, Caiolfa VR, Zamai M & Gratton E. The phasor approach to fluorescence lifetime imaging analysis. Biophysical journal 94,L14(2008). [3] Vicidomini G et al. Sharper low-power STED nanoscopy by time gating. Nature methods 8,571(2011). 1805-Plat Investigating Cellular Focal Adhesions on Nano-Patterned Substrates with Dual Color Photo-Activated Localization Microscopy Hendrik G. Deschout1, Michelle A. Baird2, Michael W. Davidson2, Joachim P. Spatz3, Aleksandra Radenovic1. 1 Institute of Bioengineering, EPFL, Lausanne, Switzerland, 2Florida State University, Tallahassee, FL, USA, 3Max Planck Institute for Intelligent Systems, Stuttgart, Germany. It is essential for cells to be able to adhere to, move within, and sense the extracellular matrix. Focal adhesions are one of the most important means through which cells achieve this goal. These are sites where trans-membrane proteins called integrins bind to specific peptides in the extracellular matrix. Inside the cells, these integrins are linked to the actin cytoskeleton through other proteins that belong to the focal adhesions. In total, there are at least 150 proteins involved in the assembly and functioning of focal adhesions [1]. This complexity, together with a size in the order of one micron or lower, strongly limits the capability of conventional imaging techniques to resolve the inner structure of focal adhesions. The recently developed single-molecule localization microscopy techniques, such as photo-activated localization microscopy, are more suitable for this purpose [2]. Another development that is currently contributing to a more detailed investigation of focal adhesions is the use of nano-engineered substrates as an extracellular matrix [3]. By precisely tuning the chemical and physical properties of such substrates, subtle changes in the behavior of focal adhesions can be provoked. Combining both approaches, we use dual color photo-activated localization microscopy [4] to investigate the co-localization between integrins and other focal adhesion proteins in cells that adhere to substrates with different nanostructured patterns of cyclic arginine-glycine-aspartic peptides [5]. References [1] Zaidel-bar et al. Nature Cell Biology 2007, 9: 858-867. [2] Tabarin et al. ChemPhysChem 2014, 15: 606-618. [3] Geiger et al. Nature Reviews Molecular Cell Biology 2009, 10: 21-33. [4] Annibale et al. Optical Nanoscopy 2012, 1:9. [5] Huang et al. Nano Letters 2009, 9: 1111-1116. 1806-Plat Quantitative Analysis of Nanoscale Lipid Bilayer Modifications via Second Harmonic Generating Probes Erick K. Moen1, Hope Beier2, Andrea Armani3, Bennett Ibey4. 1 EE-Electrophysics, University of Southern California, Los Angeles, CA, USA, 2Radio Frequency Bioeffects Branch, Human Effectiveness Directorate, Air Force Research Laboratory - Fort Sam Houston, San Antonio, TX, USA, 3Mork Family Department of Chemical Engineering and Materials Science, University of Southern California, Los Angeles, CA, USA, 4Radio Frequency Bioeffects Branch, Human Effectiveness Directorate, Air Force Research Laboratory, Fort Sam Houston, San Antonio, TX, USA. Second Harmonic Generation (SHG) microscopy is an effective tool for studying the order of symmetry-breaking interfaces, such as lipid bilayers. Recently, we have shown that disruptions in the interfacial nature of the membrane can be studied with the addition of lipophilic SHG probes, specifically Di-4ANEPPDHQ (Di-4). In addition to the conformational information provided by the SHG signal, this particular probe can be used to determine lipid phase and transmembrane voltage through the dye’s fluorescence behavior. We now strengthen our technique by changing the polarity of the incident laser beam from linear to circular polarization. The modification provides SHG signal around the circumference of the cell in every optical slice, while maintaining a sufficient signal-to-noise ratio. Utilizing nanosecond pulsed electric fields (nsPEFs) of varying pulse widths and intensities as a tool for provoking spatially and temporally minute perturbations in the cell membrane, we observe membrane poration on the nano-scale. To verify our results, we adapted a 360a Tuesday, February 10, 2015 previously-published electrical membrane model to map the theoretical angular area affected by these nsPEFs. The tests further establish the effectiveness of SHG probes, in our case Di-4, as a tool for observing subtle membrane alterations and document the extent to which electric fields of varying pulse parameters affect the cellular membrane. 1807-Plat Functional Imaging of Intact Pancreatic Islets by Inverted Selective Plane Illumination Microscopy Zeno Lavagnino, David W. Piston. Molecular Physiology & Biophysics, Vanderbilt University, Nashville, TN, USA. The islet of Langerhans is a complex aggregate of cells ~100 mm in diameter, which plays a central role in glucose homeostasis through the regulated secretion of glycolytic hormones. However, the mechanisms underlying these secretions are not yet fully understood. The behaviors of individual islet cells completely change when they are dislocated from the islet environment, so it is essential to choose experimental approaches that permit investigations in the whole islet. Fluorescence microscopy is well-suited to functional imaging in biological samples, thanks to its specificity and non-invasive quality. Selective Plane Illumination Microscopy (SPIM) has been proven to be a useful technique for imaging thick samples. It provides intrinsic optical sectioning by specifically illuminating the sample with a thin sheet of light, yielding the imaging speed of a conventional widefield microscope coupled with less photobleaching compared to confocal imaging techniques. We have developed a version of the inverted SPIM (iSPIM) [Wu et al., 2011. PNAS 108:1770813] on a widefield inverted microscope, to study the functional behavior of intact mice islets of Langerhans. By scanning the light sheet through the static sample, it is possible to obtain a volume reconstruction of the whole islet in less than 3 seconds, enabling the study of fast dynamics within the islet. Moreover, the resolution provided by a 0.8 NA objective allows for definitive measurements of individual cell’s functional details within the islet milieu. Together, the results demonstrate the multi-purpose capability of this technique. 1808-Plat Single Molecule Tracking in Living Cells - Multistep Reactions, Simulated Microscopy and New Analysis Methods Martin Linde´n, Johan Elf. Cell and Molecular Biology, Uppsala University, Uppsala, Sweden. Single particle tracking (SPT) in living cells is a quantitative and non-invasive tool for cutting through the complexity of intracellular dynamics. For example, the rates of macromolecular binding and dissociation events can be determined by monitoring the change in diffusion rates for individual labeled protein molecule that bind something relatively immobile, such as chromosomal DNA, the membrane or a ribosome. We have developed an analysis suite, vbspt.sourceforce.net ( Persson, Linde´n et al. Nat. Methods 2013), that uses a Bayesian treatment of hidden Markov models to learn the number of diffusive states and their inter-conversion rates from position trajectories of diffusing particles with random jumps in diffusion constant. However, reactions and diffusion in the living cell is more complex than the model assumptions used by vbSPT. For example, processive reactions such as transcription or translation will result in non-exponential residence times in the DNA or mRNA interacting states for the RNAP or the ribosome respectively. Furthermore, the limitations of optical imaging of rapidly diffusive molecules of finite brightness will make the position of detected molecules in each frame inaccurate and correlated between consecutive frames.To learn more about how such effects influences our analysis and limits the kind of mechanisms that can be resolved, we have developed computational tools to simulate live cell SPT, using a combination of reaction-diffusion kinetics and photophysics models. These simulations include more physical realism than the models on which vbSPT are based, which makes them suitable for optimizing experimental conditions and to improve the analysis methods. I will give a brief overview of our recent and ongoing single particle tracking projects (Hammar et al., Nature Genetics 2014, Sanamrad etal. PNAS 2014) and describe unpublished improvements in analysis methods based on simulated microscopy. 1809-Plat Mapping the Diffusive Route of Nanoparticles in Live Cells Reveals Shape to Control Nuclear Accessibility Elizabeth Hinde1, Hien T. Duong2, Bunyamin Karagoz2, Justin J. Gooding2, Cyrille Boyer2, Katharina Gaus1. 1 School of Medical Sciences, University of New South Wales, Sydney, Australia, 2Australian Centre for NanoMedicine, University of New South Wales, Sydney, Australia. Viruses have developed mechanisms of cellular entry over millions of years and shape has evolved as a critical feature to overcoming cellular barriers which restrict access to a target destination. The efficiency with which pathogens overcome cellular barriers raises the question whether drug carriers can be designed to take an ‘optimal’ route and enhance the efficacy of chemotherapeutics. Mapping molecular mobility across cellular compartments, however, has been difficult since live cell imaging has insufficient spatiotemporal resolution to follow a population of molecules in real time. Here, we used pair correlation analysis, a form of correlation spectroscopy, to map the molecular mobility of polymeric nanoparticles with millisecond and sub-micron resolution across subcellular compartments. We measured differences in nuclear accessibility for polymeric nanoparticles of different shape that correlates with doxorubicin toxicity. The latter was caused by efficient drug release in the nucleus by pathogen shaped carriers. 1810-Plat Correlative iPALM and Platinum Replica Electron Tomography Pinpoints Endocytic Proteins on the Mammalian Cell Cortex in 3D Kem A. Sochacki1, Gleb Shtengel2, Harald F. Hess2, Justin W. Taraska1. 1 National Heart Lung and Blood Institute, Bethesda, MD, USA, 2Janelia Farm: Howard Hughes Medical Institute, Ashburn, VA, USA. Super-resolution localization microscopy techniques like photoactivated localization microscopy (PALM) and stochastic reconstruction microscopy (STORM) can be used to locate proteins in cells to a resolution of 10 nm which, along with other recent super-resolution techniques, has revolutionized the field of fluorescence microscopy. However, even super-resolution fluorescence microscopy lacks the ability to see detailed structure surrounding the proteins in the way that electron microscopy (EM) has been able to do for decades. Here, we combine interferometric PALM (iPALM) with EM tomography of mammalian cell cortex platinum replicas to localize clathrinassociated proteins on clathrin coated structures in three dimensions at a resolution ~20 nm. We specifically pinpoint the position of clathrin and epsin on clathrin coated pits and find that epsin is positioned on the outer perimeter of clathrin coated plaques and along the height of the sidewalls of clathrin-coated pits. We continue to enhance this method with changes in sample preparation and we are using the method to study the nanoscale organization of membrane-associated proteins in multiple different mammalian cell lines. 1811-Plat Refsofi for Imaging Protein-Protein Interactions in Living Cells in SuperResolution Fabian Hertel1, Gary Mo1, Sam Duwe´2, Peter Dedecker2, Jin Zhang1. 1 Department of Pharmacology and Molecular Sciences, The Johns Hopkins University School of Medicine, Baltimore, MD, USA, 2Department of Chemistry, University of Leuven, Heverlee, Belgium. Virtually all cellular processes depend on networks of highly dynamic proteinprotein interactions (PPIs). Hence, deciphering cellular functions at the molecular level requires investigating the spatiotemporal characteristics of PPIs precisely within the context of these signaling networks. However, there is accumulating evidence that protein complexes are often organized into microdomains or nanodomains whose size is below the diffraction limit and thus cannot be sufficiently characterized using standard imaging techniques. Despite the tremendous progress in establishing and refining superresolution imaging techniques for fluorescently labeled proteins in live cells, observation of protein-protein interactions (PPIs) in super-resolution remains challenging. To address this problem, we developed a strategy in which PPIs induce reconstitution of different fluorescent proteins, whereupon these interactions are resolved in super-resolution using Stochastic Optical Fluctuation Imaging (SOFI) within 30 minutes after complex formation. We employed this reconstituted fluorescence based SOFI (refSOFI) to investigate the interaction between the endoplasmic reticulum Ca2þ sensor STIM1 and the pore forming channel subunit ORAI1, a process crucial for store-operated Ca2þ entry. We found that STIM1-ORAI1 interaction puncta exhibit a smaller size than that suggested by current fluorescence microscopy-based assessments. Moreover, we discovered that the density of these punctate structures is not necessarily dependent on their size. In fact, we observed that STIM1ORAI1 activation predominantly leads to the formation of new, smaller puncta instead of larger complexes. However, we identified that other perturbing factors can significantly change the size of puncta. These insights critically depend on the obtained super-resolution information. In conclusion, refSOFI represents a versatile tool to examine PPIs in living cells in superresolution. Tuesday, February 10, 2015 Platform: Cardiac Muscle Regulation 1812-Plat Overexpression of Foxo in the Heart Ameliorates Performance Decline through Enhanced UPS Processing in Aging Drosophila Anna C. Blice-Baum1, Gaurav Kaushik2, Meera C. Viswanathan1, Alexander C. Zambon3, Adam J. Engler2, Rolf Bodmer4, Anthony Cammarato1. 1 Cardiology, Johns Hopkins University, Baltimore, MD, USA, 2 Bioengineering, University of California San Diego, San Diego, CA, USA, 3 Biopharmaceutical Sciences, Keck Graduate Institute, Claremont, CA, USA, 4 Development, Aging and Regeneration Program, Sanford-Burnham Medical Research Institute, San Diego, CA, USA. Heart performance declines with age. A likely contributor to age-associated cardiac dysfunction is reduced protein quality control due to decreased function of the ubiquitin/proteasome system (UPS) and the autophagy lysosomal pathway (ALP). The transcription factor, FOXO, has been shown to be involved in the regulation of genes related to both of these interrelated pathways as well as a host of other cellular processes. Here, we investigated the effects of cardiacrestricted overexpression of dFOXO in Drosophila melanogaster, an ideal model for aging studies, exploiting the tissue-specific UAS-GAL4 expression system. Using high-speed video microscopy and motion analysis and atomic force microscopy, we showed that with age, mild heart-specific overexpression of dFOXO significantly attenuated senescence-associated cardiac functional decline and stiffening, respectively. We also determined that differing amounts of heart-specific dFOXO overexpression elicited disparate effects, the strongest driver proving fatal. Overexpression of dFOXO in all Drosophila muscle has been shown to increase lifespan, likely owing to systemic expression of autophagy related proteins and reduced ubiquitin content. Similarly, we found that dFOXO-mediated improvement in heart function with age was also accompanied by a significant decrease in ubiquitinated myocardial proteins as determined by quantitative western blot analysis. However, microarray data suggested that this reduction was caused by increased expression of genes associated with the UPS rather than autophagy, indicating that FOXO may perform its function differently in the heart than in other striated muscles. Because FOXO transcriptionally regulates genes associated with many facets of cellular life, we have established a list of specific targets that dFOXO may affect when its expression is increased to attenuate cardiac dysfunction with age. We will systematically investigate these candidates to pinpoint how mild FOXO overexpression ameliorates the natural decline in heart performance in Drosophila. 1813-Plat Obscurins’ Mechanistic Involvement in Signal Transduction at the Cardiac Intercalated Disc Maegen Ackermann, Nicole Perry, Aikaterini Kontrogianni-Konstantopoulos. University of Maryland, Baltimore, MD, USA. The intercalated disc (ID) of cardiac muscle embodies a highly ordered, multifunctional network, which is essential for the transmission of electrical stimuli and mechanical force resulting in the synchronous contraction of the heart. Recently, a plethora of proteins have been identified as novel components of the ID. The challenge now lies in their characterization. Here we focus on the molecular and functional description of two novel members of the ID, obscurin-80 and obscurin-40. Obscurins are a family of proteins expressed in striated muscles where they localize to distinct subdomains. The members of the obscurin family are multidomain proteins composed of adhesion modules and signaling domains, resulting from extensive alternative splicing of transcripts arising from the single OBSCN gene. Recent work from our laboratory has demonstrated that complex splicing at the 3’ end of the obscurin transcript gives rise to at least two novel obscurins, obscurin-80 (obsc-80) and obscurin-40 (obsc-40), named after their predicted molecular weights. Using immunofluorescence and immunoelectron microscopy, we show that obsc-80 and obsc-40 localize to the ID of developing and adult murine cardiomyocytes. Using biochemical assays we further demonstrate that both obsc-80 and obsc-40 exist in a complex with major ID proteins, including N-cadherin, connexin-43, vinculin, and ankyrin-G. The PH domain present in both obsc-80 and obsc-40 binds specifically and directly binds to phosphatidylinositol 3,4 and 4,5 bisphosphates, likely targeting both proteins to the ID membrane. Overexpression of the obscurin PH domain results in decreased phosphorylation of Akt, therefore reducing Akt activation. This suggests a potential role for obsc80 and obsc-40 in the regulation of cell growth and proliferation via the Akt pathway. Further experiments are underway to examine the functional activities of obsc-80 and obsc-40 at the ID and their regulation in health and disease. 361a 1814-Plat The N-Terminal Hypervariable Region of Troponin T Differentially Modulates the Affinity of Tropomyosin-Binding Sites Chinthaka K. Amarasinghe, Jian-Ping Jin. Physiology, Wayne State University School of Medicine, Detroit, MI, USA. The troponin complex plays a central role in the allosteric function of sarcomeric thin filaments by enacting conformational changes during the Ca2þ-regulated contraction and relaxation of striated muscle. The troponin subunit T (TnT) has two binding sites for tropomyosin (Tm) and is responsible for anchoring the troponin complex to the thin filament. Although the C-terminal and middle regions of the TnT polypeptide chain are highly conserved among the three muscle type isoforms, the hypervariable N-terminal region has evolutionarily diverged significantly among isoforms. Previous studies have shown that the N-terminal variable region fine-tunes Ca2þ regulation of muscle contractility via modulation of the overall molecular conformation of TnT, and its interactions with Tm. In the present study, we engineered intact TnT and representative fragments of TnT, expressed them in E. coli, and prepared purified proteins for functional studies. Tropomyosin binding affinity was analyzed using affinity chromatography and solid phase protein binding assays to investigate the modulatory effects of the N-terminal variable region. The results demonstrated that in the absence of the N-terminal variable region, TnT’s conserved middle region and C-terminal T2 region Tm-binding sites showed comparable Tm-binding affinities across isoforms. The data demonstrate that without the modulatory effect of the N-terminal variable region, the intrinsic Tm-binding affinities of the two sites are both high. In contrast, the presence of the isoform specific N-terminal variable region differentially reduces the binding affinity of TnT for Tm, primarily at the middle region binding site. These novel findings indicate that the N-terminal variable region plays a key role in the functional difference of muscle fiber type-specific, developmental, splice variant, and pathogenic TnT isoforms by modulating the interactions with Tm during the contraction and relaxation of cardiac and skeletal muscle. 1815-Plat Constitutive Phosphorylation of Myosin Regulatory Light Chain (RLC) in vivo is Maintained by Low Kinase and Phosphatase Activities Audrey N. Chang1, Patrick M. Cowley2, Anthony J. Baker2, Kristine E. Kamm1, James T. Stull1. 1 Physiology, University of Texas Southwestern Medical Center, Dallas, TX, USA, 2Cardiology, VA Medical Center & Univ. Calif. San Francisco, San Francisco, CA, USA. The importance of RLC phosphorylation in enhancing cardiac myofibrillar contraction is well-known. In anesthetized mice the extent of RLC phosphorylation (45%) was not changed by changes in sympathetic tone with prolonged infusion of the beta-agonist dobutamine or treatment with the beta-blocker propranolol. The goal of this study was to determine if the constitutive RLC phosphorylation in vivo was limited to half of the RLC due to: a) negative cooperativity for phosphorylation of two heads in myosin; or b) due to a steric constraint in myofibrils blocking access of soluble cardiac myosin light chain kinase (cMLCK) to RLC. We measured the kinetic properties of RLC phosphorylation in native myosin filaments and myofibrils. Results showed that RLC was phosphorylated by a pseudo-first order rate in both preparations with maximal phosphorylation over 90%. Thus, RLC for each head in myosin was readily available for phosphorylation. Pacing trabeculae at 1.5 Hz for 30 minutes increased RLC phosphorylation from 2051 % to 4353 %. Consistent with biochemical results, RLC phosphorylation increased to 9153 % when myosin light chain phosphatase activity was inhibited with calyculin A while pacing. Together, these results exclude negative cooperativity and steric blocking as mechanisms limiting RLC phosphorylation. We determined that the heart has a high cMLCK content (2.450.1 mM) compared to the MLCK present in fast skeletal muscle (0.550.03 mM), but similar to the MLCK content in smooth muscle (3.450.2 mM). However cMLCK has a low specific activity compared to the other MLCKs. In conclusion, the extent of RLC phosphorylation in a normally beating heart is limited by cMLCK with its low activity in balance with low myosin light chain phosphatase activity. RLC phosphorylation is insensitive to sympathetic activation or inhibition in vivo. 1816-Plat Epigallocatechin-3-Gallate Reverses the Defects in Modulation of Ca2DSensitivity by Troponin I Phosphorylation Caused by Hypertrophic and Dilated Cardiomyopathy Mutations in Cardiac Muscle Maria Papadaki, Petr Vikhorev, Steven Marston, Andrew Messer. NHLI, Imperial College, London, United Kingdom. Heart muscle contraction is regulated via the b-adrenergic response that leads to phosphorylation of Troponin I (TnI) at Ser 22/23, which changes the Ca2þsensitivity of the cardiac myofilament. Previously it has been shown that mutations found in Dilated Cardiomyopathy (DCM) and Hypertrophic 362a Tuesday, February 10, 2015 Cardiomyopathy (HCM) patients abolish the relationship between TnI phosphorylation and Ca2þ-sensitivity (uncoupling). Ca2þ-sensitisers and Ca2þ-desensitisers that act upon troponin alter the Ca2þsensitivity of the myofilament but their relationship with TnI phosphorylation has never been studied before. Epigallocatechin-3-gallate (EGCG) is a major extract of green tea and it also acts as a Ca2þ-desensitiser by binding to Troponin C of the myofilament. 100 mM EGCG decreased Ca2þ-sensitivity of phosphorylated and unphosphorylated wild-type thin filaments equally (by 2.1550.45 and 2.8050.48fold respectively), retaining the coupling. In contrast, EGCG reduced Ca2þ-sensitivity of phosphorylated but not unnphosphorylated thin filaments containing 8 DCM (TPM1 E54K and E40K, TNNC1 G159D, TNNI3 K36Q, ACTC E361G) and HCM (TPM1 E180G, TNNT2 K280N, ACTC E99K)causing mutations. As a result the dependence of Ca2þ-sensitivity upon TnI phosphorylation of uncoupled mutant thin filaments was restored in every case. In single myofibrils, EGCG reduced Ca2þ-sensitivity of force and kACT and also preserved the coupling with wild type and restored coupling with ACTC E361G mutant myofibrils. The effect of EGCG demonstrates that it is possible to reverse the pathological defects in troponin caused by HCM mutations pharmacologically. 1817-Plat Myosin-Binding Protein C Corrects an Intrinsic Non-Uniformity in Cardiac Excitation-Contraction Coupling Michael J. Previs1, Benjamin L. Prosser2, Ji Young Mun3, Samantha Beck Previs1, James Gulick4, Kyounghwan Lee3, Jeffrey Robbins4, Roger Craig3, W. Jonathan Lederer5, David M. Warshaw1. 1 Department of Molecular Physiology and Biophysics, University of Vermont, Burlington, VT, USA, 2Department of Physiology, University of Pennsylvania, Philadelphia, PA, USA, 3Department of Cell and Developmental Biology, University of Massachusetts Medical School, Worcester, MA, USA, 4Department of Pediatrics and the Heart Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA, 5 Department of Physiology, University of Maryland School of Medicine, Baltimore, MD, USA. Cardiac contraction is initiated by calcium release from the sarcoplasmic reticulum through channels (ryanodine receptors) that are located near the sarcomere ends. Once released, calcium must diffuse towards the sarcomere center to fully activate the actomyosin contractile system. This physical arrangement should lead to a spatial calcium gradient and thus non-uniform contractile activation. We hypothesize that myosin-binding protein C (MyBP-C), a potent thin filament activator, is localized to the sarcomere center (C-zone) to mitigate the potential deficit in calcium activation. We used EM and super-resolution STORM microscopy to visualize the relative positions of the ryanodine receptors and MyBP-C within the sarcomere. Laser scanning confocal microscopy of calcium transients in isolated cardiac cells showed that calcium concentrations at the center of each sarcomere lagged those near the ends of each sarcomere by as much as 150 nM. The functional impact of this calcium gradient was determined by examining the sliding of native, calcium-sensitive actin-thin filament shards over native mouse cardiac myosin-thick filaments using a TIRFM-based motility assay. The presence of MyBP-C enhanced the fraction of thin filaments moving within the thick filament C-zone (pCa50 6.5 5 0.04 vs 6.4 5 0.02; p<0.01). 3D EM reconstructions of native thin filaments suggest this calcium sensitization results from MyBP-C’s N-terminal domains shifting tropomyosin on the thin filament. Using an analytic model, we show that MyBP-C residing within the C-zone can counterbalance differences in calcium activation within the sarcomere during the early phase of contraction. Thus, MyBP-C’s localization to the C-zone may function to correct an intrinsic defect in cardiac excitation-contraction coupling, and any disturbance of MyBP-C localization or function will contribute to the consequent cardiac pathologies. 1818-Plat Direct Detection of the Thermodynamics and Structural Kinetics of a 2-Color SERCA Biosensor by Transient Time-Resolved FRET Simon J. Gruber1, Rebecca Goldblum2, Jenica Zhong2, Kurt Peterson2, Tory M. Schaaf2, Joseph M. Autry2, Gregory D. Gillispie2, David D. Thomas2, Joseph M. Muretta2. 1 Mount Sinai Hospital, New York, NY, USA, 2Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, USA. We are investigating the structural dynamics of the calcium pump SERCA, using a 2-color biosensor and time-resolved FRET measurements. SERCA couples ATP hydrolysis to Ca2þ transport across the sarcoplasmic or endoplasmic reticulum membrane. Crystal structures suggest that SERCA structure changes dramatically during active Ca2þ transport, including large movements in the cytoplasmic N and A domains, coupled to more subtle changes in the transmembrane domain. However, there is little direct evidence showing how changes in SERCA structure coordinate, in solution and in cells, with individual steps in the pump’s ATPase and Ca2þ transport cycles. Furthermore, SERCA is a viable therapeutic target for a host of diseases_heart failure, diabetes, and cancer_so it is important to understand how the ATPase and Ca2þ transport cycles are coupled to changes in the protein’s structure. We are addressing these questions using a recombinant 2-color SERCA biosensor (RFP and GFP fused to the N and A domains) developed by Robia and coworkers for live-cell work. Here we use it in isolated membrane fragments to detect changes in SERCA structure in response to changes in Ca2þ and nucleotide concentrations, both at equilibrium and in the transient phase after rapid mixing. Our results: 1) reveal critical changes in the structural-dynamics of the SERCA cytoplasmic domains upon Ca2þ and nucleotide binding; and 2) suggest a structural-kinetic mechanism for Ca2þ activation of ATPase cycling with coordinated changes in the position of the cytoplasmic domains in association with sequential binding of Ca2þ ions and ATP. 3) These results also allow direct testing of x-ray crystal structure models in solution and in living cells and inform the discovery of novel SERCA modulators that directly target the pump’s structural dynamics. 1819-Plat Phospholamban-Independent Adrenergic Reserve in SERCA2 Ablated Hearts Frazer I. Heinis1, Joseph M. Metzger2. 1 Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN, USA, 2Integrative Biology and Physiology, University of Minnesota, Minneapolis, MN, USA. Normal cardiac contraction and relaxation requires efficient operation of the SR Ca2þ ATPase, SERCA2a. In failing hearts, decreased SERCA2a expression and slowed SR Ca2þ reuptake are thought to significantly underlie contractile dysfunction. We have studied the relationship between decreased SERCA2a expression and cardiac function using a mouse model of conditional cardiac Serca2 ablation, the Serca2fl/fl mouse. In this model, Cre activation by tamoxifen injection efficiently deletes Serca2 from the heart; four weeks postknockout, cardiac SERCA2a protein content is below 5% of normal levels. Serca2 KO mice survive 7-10 weeks post knockout with only mild in vivo impairment prior to this time, suggesting that this loss of SERCA2a protein can be temporarily compensated. We found that isolated SERCA2a KO hearts retained the ability to respond to ß-adrenergic stimulation ex vivo despite the loss of most SERCA2a protein. Because the protein regulator of SERCA2a, phospholamban (PLN), is thought to be a major component of the cardiac ßadrenergic response, this finding was unexpected and warranted further detailed investigation. To identify the mechanism underlying this preserved ß-adrenergic response in SERCA2-depleted hearts, we bred Serca2fl/fl and PLN-/mice to generate an inducible Serca2 knockout mouse line lacking PLN. Serca2fl/fl;PLN-/- mice were injected with tamoxifen to induce Serca2 gene disruption, and isolated hearts from Serca2KO;PLN-/- (‘‘DKO’’) mice were evaluated by Langendorff perfusion 4-5 weeks after Serca2 knockout. Control PLN-/- hearts show little response to ß-adrenergic stimulation, and DKO hearts have impaired contractility under baseline conditions. DKO hearts, however, show improved systolic and diastolic function when perfused with 50 nM isoproterenol, indicating that targets of ß-adrenergic signaling beyond PLN are able to support a significant inotropic and lusitropic response when cardiac contractility is impaired. We will discuss how these findings support new mechanisms underlying heart performance during stress and disease. Platform: Protein Dynamics and Allostery II 1820-Plat Conformational Dynamics of Single HIV-1 Envelope Proteins on the Surface of Native Virions James B. Munro1, Jason Gorman2, Xiaochu Ma3, Zhou Zhou4, James Arthos5, Dennis Burton6, Wayne Koff7, Joel Courter8, Amos Smith8, Peter Kwong2, Scott Blanchard4, Walther Mothes3. 1 Molecular Biology and Microbiology, Tufts University, Boston, MA, USA, 2 Vaccine Research Center, National Institutes of Health, Bethesda, MD, USA, 3Microbial Pathogenesis, Yale University, New Haven, CT, USA, 4 Physiology and Biophysics, Weill Cornell Medical College, New York, NY, USA, 5Laboratory of Immunoregulation, National Institutes of Health, Bethesda, MD, USA, 6Immunology and Microbial Science, Scripps Research Institute, La Jolla, CT, USA, 7International AIDS Vaccine Initiative, New York, NY, USA, 8Chemistry, University of Pennsylvania, Philadelphia, PA, USA. The HIV-1 envelope (Env) mediates viral entry into host cells. While static images of Env in unliganded and ligand-bound forms define distinct Tuesday, February 10, 2015 conformations, direct observations of Env dynamics have yet to be realized. Here we apply single-molecule fluorescence resonance energy transfer (smFRET) imaging to elucidate the dynamics of native Env trimers on the surface of HIV-1 virions. Our observations indicated that unliganded HIV-1 Env transitions between three distinct pre-fusion conformations, which are affected by the viral receptor and co-receptors. Differences in conformational dynamics and ligand responsiveness of neutralization-sensitive and neutralizationresistant HIV-1 isolates delineated a dynamics-based mechanism of immune evasion. Broadly neutralizing antibodies stabilized one distinct pre-fusion conformation of Env, indicating the importance of the observed dynamics to HIV-1 Env function. 1821-Plat Allosteric Regulation of Nipah Virus Entry into Host Cells Sameer Varma, Priyanka Dutta, Mohsen Botlani. Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, FL, USA. Nipah viruses are highly virulent and cause recurring encephalitis in humans with 77% mortality. The entry of these viruses into host cells is triggered when specific glycoproteins on the viral membrane, called attachment proteins, bind to their appropriate receptors on the host cell membrane. The attachment proteins have separate domains for receptor binding and mediating virus-host membrane fusion. However, the molecular details of how the receptorbinding signal transduces from the receptor-binding domain to the fusionmediating domain remains unknown. Understanding this process has been challenging mainly because receptor binding induces only minor structural changes in the receptor binding domain (mean deviation < 0.2 nm). This implies that signal transduction occurs primarily via changes in side-chain rotations and fluctuations. Consequently, to understand signaling in such scenarios, one needs to look beyond examining differences between two protein structures. An understanding of signal transduction in such systems requires a quantitative assessment of differences in structural ensembles. Here we will present the development of new methods to quantitatively evaluate differences in conformational ensembles [1,2]. The primary challenge that these methods overcome is associated with comparing two high dimensional vector spaces. In addition, we will present how we have used this method in conjunction with accelerated conformational sampling techniques to illuminate the molecular details underlying the allosteric regulation of Nipah entry into host cells. These studies highlight, in general, how signals can be transferred across nanometer long distances in proteins without major backbone rearrangements. We anticipate that our method and approach will be applicable to several other systems where allosteric signaling is achieved via small changes in protein structure. [1] RE Leighty, S Varma, J. Chem. Theory Comput. 9 (2013) 868. [2] S Varma, M Botlani, RE Leighty, Proteins (2014) In press. 1822-Plat Specific Protein-Lipid Interactions Stabilize an Active State of the Beta 2 Adrenergic Receptor Chris Neale1, Henry D. Herce1, Re´gis Pome`s2, Angel E. Garcı´a1. 1 Rensselaer Polytechnic Institute, Troy, NY, USA, 2Hospital for Sick Children, Toronto, ON, Canada. Intercellular communication is essential for many facets of multicellular life. To accomplish this communication, the human genome contains over 800 G protein-coupled receptors (GPCRs), which have evolved to bind thousands of different chemicals and evoke varied cellular responses. The beta 2 adrenergic receptor (B2AR) is a well-studied GPCR that mediates the fight-or-flight response and is the target of sixteen approved drugs. However, the stepwise mechanism by which extracellular ligand binding leads the B2AR to activate an intracellular G protein remains unclear. Moreover, molecular dynamics (MD) simulations indicate that, in the absence of an intracellular binding partner, activated forms of the receptor are unstable and undergo deactivation on the microsecond timescale. To understand the source of this instability, we conduct extensive (0.25 millisecond) MD simulations of two forms of the active state of the B2AR and define conditions that are sufficient to prolong receptor activity. The influence of lipid composition on receptor activity identified in these simulations is corroborated by comparison to experiment. This research was supported by NSF MCB-1050966. 1823-Plat Dynamics of M2 Proton Channel: Insights into the Motions of the Primary and Secondary Gates Joana Paulino1, Ivan Hung2, Timothy A. Cross1,2. 1 FSU, Tallahassee, FL, USA, 2NHMFL, Tallahassee, FL, USA. M2 proton channel is essential for Influenza A life cycle. The antiviral drug amantadine (AMT) used to block the M2 channel prior to the recent S31N mutation. Based on the abundant structural information on M2 we have char- 363a acterized backbone and side-chain motions for Trp41 and Val27, in constructs of the transmembrane domain (M2TM) and the full length M2 protein (M2FL) reconstituted in lipid bilayers. M2 proton permeation has been shown to be dependent on the His37-Trp41 cluster, where Trp41 is the primary gate. Proton conductance directionality is lost upon Trp41 mutation. We have characterized the global rotation motion of M2TM using 2H ssNMR. Interestingly, M2TM and M2FL have very similar dynamics for the backbone and side-chain of Trp41, as indicated by the 15N powder spectra of the Trp41 site labeled in both constructs. The collapsed powder spectra of 15NεTrp41 in M2FL and M2TM indicate that Trp41 side chain undergoes motion on a fast time scale. The separated local field spectrum of aligned M2TM indicates that Trp41 side-chain is undergoing large amplitude motion that broadens the 15Nε peak. The mean orientation for the Trp41 is being determined for M2TM and M2FL, moreover the effect of AMT binding and pH activation is being addressed. We also derived the motional model of Val27 side-chain, considered a secondary gate for M2 and essential for AMT inhibition on the wild type protein. The side-chain of Val27 undergoes a two site jump motion about the Ca-Cb bond, with unequal populations. The addition of AMT induced line broadening and a significant increase in T2 (T2apo ¼ 27 þ- 7 ms T2drug¼43 þ- 3 ms). The spectra of the same site on the mutant protein (M2TM_S31N-d8Val27) showed no changes upon addition of Amt. 1824-Plat Identification of an Endogenous Allosteric Modulator’s Binding Site at the Human Cannabinoid-1 Receptor, Using Forced-Biased Metropolis Monte Carlo Simulated Annealing Method (MMC) and Molecular Dynamics Derek M. Shore, Dow P. Hurst, Diane L. Lynch, Patricia H. Reggio. Chemistry and Biochemistry, UNC Greensboro, Greensboro, NC, USA. The CB1 endogenous, positive allosteric modulator, lipoxin A4, increases the equilibrium binding and efficacy of CP55,940 and anandamide (orthosteric agonists), yet has no significant effect when applied alone. We have reported that ORG27569 (a negative CB1 allosteric modulator) binds in the THM3/6/7 region (Shore et al., JBC, 2013); here, ORG27569 sterically blocks movements of the second and third extracellular (EC) loops, as well as those of TMH6, that are necessary for G protein-mediated signaling. Because lipoxin A4 is a positive allosteric modulator, one would not expect it to sterically block these functionally-important conformational changes. To identify lipoxin A4’s binding site(s) at CB1, we used the Forced-Biased Metropolis Monte Carlo simulated annealing program, MMC. In this method, lipoxin A4 was separated into 4 fragments. Four MMC runs were performed, in which our in silico CB1 receptor model (with CP55,940 docked) was immersed in a box filled with copies of one of these fragments. The system chemical potential was then systematically annealed, causing only those fragment copies with the best free energy of binding to the protein to remain. MMC results were used as a starting point for Glide automated-docking studies of lipoxinA4. Molecular dynamics simulations were also performed to study how lipoxin A4 may enter CB1. Here, CB1 was placed in a fully hydrated, POPC bilayer; 14 lipoxin A4 molecules (7 per leaflet) were placed with random orientations, around the receptor. Altogether, these results suggest that lipoxin A4 may bind in the TMH3/6/7, extending extracellularly. Lipoxin A4 may act as a positive allosteric modulator by forming electrostatic interactions with the EC-1 and EC-3 loops, promoting an active loop conformations. [Support: RO1 DA003934 and KO5 DA021358 (PHR)] 1825-Plat Ligand-G Protein Allosteric Communication through Internal Waters in GPCR Complexes Roman Osman1, Jose Carlos Gomez2, Mihaly Mezei1, Dov Barak1, Arnau Cordomi2, Leonardo Pardo2. 1 Structural and Chemical Biology, Mount Sinai School of Medicine, New York, NY, USA, 2Laboratori de Medicina Computacional, Universitat Auto`noma de Barcelona, Barcelona, Spain. Numerous structures of GPCRs show that the changes in the ligand pocket are small compared to the much larger changes in the G protein site. This challenges the notion that agonist binding induces changes in the protein that lead to G protein binding and receptor activation, which is reflected in the thermodynamics of the linked allosteric effect of agonist binding on G protein affinity. A possible involvement of internal waters, present in all GPCRs, could provide a mechanism for the connection between the ligandbinding pocket and the G protein-binding site. Using MD simulations and an enhanced inhomogenous fluid solvation theory we obtain the free energy of the internal waters and their contribution to the dynamics of protein complexes. We focus on the Adenosine-2A receptor in complex with antagonist, agonists and in ternary complex with a G protein modeled by a nanobody. 364a Tuesday, February 10, 2015 Graph theoretical analysis of the network defined by inter-water H-bonds shows that residues in Hx3 and Hx7 (L3.43, N7.49 and Y7.53) interrupt the network creating two isolated clusters – one large encompassing the ligand binding pocket and a small one in the G protein site – preventing inter-site communication. Inclusion of protein sites that H-bond waters establishes a continuous but very weak connectivity between the isolated clusters in the complex with an antagonist. In contrast, the presence of an agonist creates multiple pathways and increases network connectivity by ca. 30fold, establishing an allosteric link between the ligand and G protein. We are currently investigating network connectivity in the ternary complex agonist,receptor,Gprotein. 1826-Plat Insertion of b-Barrel Proteins in Gram-Negative Bacteria Karl Lundquist, James C. Gumbart. Physics, Georgia Institute of Technology, Atlanta, GA, USA. Gram-negative bacteria possess two membranes, the inner and outer of which contain primarily a-helix and b-barrel proteins respectively. In recent years, significant progress has been made in understanding insertion and assembly of proteins into the inner membrane, while the same process in the outer membrane has remained elusive. In 2013, the crystal structure of BamA, the central and essential component of the b-barrel assembly machinery (BAM), was released, paving the way for rapid progress in understanding the insertion and assembly process. All-atom molecular dynamics simulations have been performed, revealing many novel features including lateral gate opening between the first and last barrel strands, and a significantly thinner, destabilized membrane region near the putative insertion site. However, many questions remain, including the role of the periplasmic domains, the mode of substrate recognition, and the energetic factors driving function in the absence of both ATP and an electrochemical gradient. We have performed novel equilibrium simulations of the protein in its native lipopolysaccharide environment including its essential periplasmic domain. Here, we present a comparison of free energy associated with lateral-gate opening for native systems, as well as systems with strand modifications and augmentations which yield insight into driving energetics and substrate recognition. 1827-Plat Super-Resolution Mapping of the Dynamics of Periodic Structural Defects in Collagen Fibrils Andrew Dittmore1, Jonathan Silver1, Barry Marmer2, Gregory I. Goldberg3, Keir C. Neuman1. 1 Laboratory of Molecular Biophysics, National Institutes of Health, Bethesda, MD, USA, 2Department of Biochemistry and Molecular Biophysics and Division of Dermatology,, Washington University School of Medicine, St Louis, MO, USA, 3Department of Biochemistry and Molecular Biophysics and Division of Dermatology,, Washington University School of Medicine, St.louis, MO, USA. Single-molecule tracking of matrix metalloproteinases (MMPs) moving on fibrillar collagen reveals a regular binding pattern with a 1.1 mm periodicity. The binding sites exhibit collective motion that preserves the distribution but not the phase of the binding pattern. Over short timescales (~1 s), the motion of individual binding sites is consistent with diffusion in a harmonic potential. At longer timescales (~20 s), the potential wells slowly migrate, bifurcate, and merge. The dynamic nature of the binding sites suggests that they correspond to transient local defects in the collagen fibril structure. However, the long-range order of their pattern, exceeding any known structural scale of the fibril, indicates a collective defect formation process. We propose a model in which internal strain energy in fibrillar collagen is relieved by the formation of defects that are distributed along its length. This model falls into the general class of mechanical instabilities that generate long-range spatial patterning in physical systems ranging from mud cracking to skin wrinkling. However, unlike cracks and wrinkles that are stable structures, the microscopic fibril features thermally excited structural dynamics and self-healing of defect states. One physiological consequence of the proposed model is that external tension opposing the internal strain in the fibril can suppress defect formation and exposure of the MMP binding sites. Experiments showing that external loading attenuates the enzymatic degradation of fibrillar collagen are consistent with this prediction of the model. More generally, many aspects of collagen degradation, including cleavage initiation, processivity, and kinetics, may largely be a consequence of a previously unrecognized structural heterogeneity in the underlying fibrillar substrate. Thus, mapping the periodic array of defects in the molecular architecture of collagen elucidates a key feature regulating enzymatic activity and remodeling of the extracellular matrix. Platform: Systems Biophysics 1828-Plat Symmetry and Scale Orient Min Oscillation Patterns in Bacterial Shape Sculptures Fabai Wu, bas van Schie, Juan Keymer, Cees Dekker. Bionanoscience, Delft University of Technology, Delft, Netherlands. What are the effects of cellular confinement and cell shape on the intracellular organization, and vice versa? Here we present a novel nanostructuring technique to systematically ‘sculpt’ living bacterial cells into defined shapes, e.g. squares and rectangles, to explore the spatial adaptation of Min proteins that oscillate pole-to-pole to assist division-site selection at the mid-plane in rodshape Escherichia coli. In a wide geometric parameter space, spanning cell volumes from 2x1x1 to 11x6x1 mm3, Min proteins are found to exhibit versatile oscillation patterns, sustaining rotational, longitudinal, diagonal, stripe, and even transversal modes. Their quantitative distributions reveal two essential properties that orient the Min patterns, viz., aligning to symmetry axes and a characteristic length range of 3-6 mm. Within this range, Min pattern are found to scale in adaptation to the cell length. The finding that Min patterns directly capture the symmetry and scale of the cell boundary to spatially regulate cell division refutes all previous geometry-sensing models that were based on the longest distance, membrane area or curvature. Using numerical simulations, we find that global symmetry selection and gradient scaling both derive from the local microscopic self-activation and inhibition kinetics, which are key components of the Turing reaction-diffusion mechanism underlying Min oscillatory dynamics. Both geometry-sensing properties only emerge within fully three-dimensionally confined volumes, contrasting in vitro Min waves on planes and theories with a fixed wavelength. Our results show that simple molecular interactions are capable of bridging the characteristics of cell shape to the spatiotemporal regulation of essential cellular processes. 1829-Plat Quantitative Analysis of RNA Interference by mRNA Couting at Single-Cell Level Hye Ran Koh, Sua Myong. UIUC, Urbana, IL, USA. RNA interference (RNAi) is a gene regulation pathway induced by small RNA molecules, microRNA (miRNA) and small interfering RNA (siRNA). It is a multi-step process consisting of the small RNA genesis by Dicer, guide strand selection and the guide strand-mediated gene silencing either through mRNA cleavage or translation inhibition by RNA-induced silencing complex (RISC). Even though RNAi is one of the major techniques to regulate gene, the mechanistic details of each step are yet to be determined. Especially, unveiling which step is the rate-determining step would deepen the understanding about the molecular mechanism of RNAi and could contribute designing more efficient small RNAs for gene silencing. To address the question, the quantitative analysis of RNAi is required, which benefits from the detection of single mRNA at single-cell level. Here, we incorporated single-molecule fluorescence in situ hybridization (smFISH) to count mRNA at single-cell level and screened multiple sets of small RNAs which mimic miRNA, siRNA, pre-miRNA and pre-siRNA. Together with single-molecule fluorescence resonance energy transfer (smFRET) which enables us to quantify the RNA cleavage by Dicer, we discovered that the cleavage step by Dicer controls the overall silencing kinetics. Interestingly, we also found that Dicer is sensitive to 3’ overhang not RISC. The quantitative analysis of RNAi using smFISH together with smFRET would be a powerful platform to study RNAi and other gene-regulating system. 1830-Plat Environmental Statistics and Optimal Regulation David A. Sivak1, Matt Thomson2. 1 Physics, Simon Fraser University, Burnaby, BC, Canada, 2Systems and Synthetic Biology, University of California, San Francisco, San Francisco, CA, USA. Any organism is embedded in an environment that changes over time. The timescale for and statistics of environmental change, the precision with which the organism can detect its environment, and the costs and benefits of particular protein expression levels all will affect the suitability of different strategies such as constitutive expression or graded response - for regulating protein levels in response to environmental inputs. We propose a general framework here specifically applied to the enzymatic regulation of metabolism in response to changing concentrations of a basic nutrient - to predict the optimal regulatory strategy given the statistics of fluctuations in the environment and measurement apparatus, respectively, and the costs associated with enzyme production. We Tuesday, February 10, 2015 use this framework to address three fundamental questions: (i) when a cell should prefer thresholding to a graded response; (ii) when there is a fitness advantage to implementing a Bayesian decision rule; and (iii) when retaining memory of the past provides a selective advantage. We specifically find that: (i) relative convexity of enzyme expression cost and benefit influences the fitness of thresholding or graded responses; (ii) intermediate levels of measurement uncertainty call for a sophisticated Bayesian decision rule; and (iii) in dynamic contexts, intermediate levels of uncertainty call for retaining memory of the past. Statistical properties of the environment, such as variability and correlation times, set optimal biochemical parameters, such as thresholds and decay rates in signaling pathways. Our framework provides a theoretical basis for interpreting molecular signal processing algorithms and a classification scheme that organizes known regulatory strategies and may help conceptualize heretofore unknown ones. 1831-Plat Fundamental Constraints on the Abundances of Chemotaxis Proteins Anne-Florence Bitbol, Ned S. Wingreen. Lewis-Sigler Institute, Princeton University, Princeton, NJ, USA. Flagellated bacteria, such as Escherichia coli, perform directed motion in gradients of concentration of attractants and repellents in a process called chemotaxis. Transmembrane chemoreceptors, which bind attractants and repellents, control the activity of the histidine kinase CheA, which phosphorylates the cytoplasmic response regulator CheY. Phosphorylated CheY binds to FliM in the flagellar motors. This binding controls the direction of the rotation of the motors, and hence the motion of the cell. E. coli chemotaxis is a model for signal transduction. However, this signaling system features very large abundances of the proteins involved, compared to those of analogous or homologous systems. Estimating the timescales of the pathway enables us to trace the need for this large number of chemotaxis proteins to the specific requirement of the chemotaxis system for fast response. We also show that further constraints arise from the requirements of self-assembly, both of flagellar motors and of chemoreceptor arrays. A surprising fact is that the abundance of all the chemotaxis proteins significantly increases in poorer medium, while their proportions are conserved. Artificially over-expressing chemotaxis proteins in a concerted manner has been shown to increase chemotactic efficiency. Employing a chemotaxis pathway model, we show that the gain of the pathway at the level of the response regulator CheY increases upon concerted over-expression of the chemotaxis protein abundances. This increase of the gain could allow cells to become sensitive to even smaller changes of concentrations of attractant, which may be beneficial in poor nutritional conditions. Besides, over-expression yields higher cooperativity of receptor teams, which could further contribute to increasing the sensitivity of the pathway. We also demonstrate that physiological proportions yield near-optimal gain, and that the pathway is robust to variations in abundance of the motor protein FliM. 1832-Plat Early Lineage Bifurcation during Differentiation of Embryonic Stem Cells Revealed by Single-Cell Transcriptomics Stefan Semrau1,2, Johanna Goldmann1, Magali Soumillon3, Tarjei Mikkelsen3, Rudolf Jaenisch1, Alexander van Oudenaarden2. 1 Whitehead Institute for Biomedical Research, Cambridge, MA, USA, 2 Hubrecht Institute, Utrecht, Netherlands, 3Broad Institute of MIT and Harvard, Cambridge, MA, USA. The development of more efficient and specific in vitro differentiation protocols is hampered by the inherently heterogeneous cellular response to lineage specifying signals. Here, we used our recently developed single-cell RNAseq method [1] and single-molecule FISH [2,3] to quantify the variability in transcriptional states of thousands of individual mouse embryonic stem cells during differentiation with retinoic acid. After a fast initial response cells exhibited delayed commitment to differentiation followed by a bifurcation into an ectoderm-like and an extraembryonic endoderm-like transcriptional state. Notably, many cells assumed an extraembryonic endoderm-like state through an ectoderm-like intermediate. In contrast to the current belief that cells are initially refractory to lineage specifying signals we found that an early, short retinoic acid pulse was able to influence the lineage decision while the cells were not yet committed. Additionally, gene expression variability, as quantified by the entropy per gene, showed a non-trivial dynamical behavior: while the accepted paradigm states that the entropy of the whole system should decrease monotonically during differentiation, we found that the average entropy per gene first decreased before it increased towards its global maximum at the end of the differentiation time course. Interestingly, this variability ‘‘bottleneck’’ coincided with the point of commitment beyond which cells were not able to return to their pluripotent initial state. In conclusion, our study demonstrated that the first few hours of a differentiation protocol can be critical for the 365a lineage decision. This observation is vital for the scientific community interested in improving differentiation protocols. [1] Soumillon, Cacchiarelli, Semrau et al., 2014, biorxiv: http://dx.doi.org/ 10.1101/003236 (under review) [2] Raj et al., Nat Methods, 2008, vol. 5 (10), pp. 877-879 [3] Semrau et al., Cell Reports, 2014, vol. 6 (1), pp. 18-23 1833-Plat Emergent Behaviours of Stem Cells in Organogenesis Demonstrated by Hybrid Modelling Benjamin A. Hall1, Nir Piterman2, Alex Hajnal3, Jasmin Fisher1. 1 Microsoft Research, Cambridge, United Kingdom, 2Department of Computer Science, University of Leicester, Leicester, United Kingdom, 3 Institute of Molecular Life Sciences, University of Zurich, Zurich, Switzerland. The development of organs from stem cells is a complex process determined by spatial and temporal control mechanisms, to create well defined and complex 3D structures. In this process, a homeostasis between the processes of cell division and cell death is required. Executable biological models describe signalling and developmental processes at a high level of abstraction, allowing for mechanistic behaviour to be modelled in the absence of precise kinetic information. However, the physical process of 3D growth in the development of an organ is harder to abstract. Here we present a hybrid model of the development of the C. elegans germline from a pool of stem cells. The model combines a description of signalling and developmental processes using an executable model, developed in the BioModelAnalyzer tool (http://biomodelanalyzer. research.microsoft.com/), with a physical model of cell interactions described using Brownian dynamics simulation. This tool uniquely allows us to study the dynamics of organ development and growth, and here we will present new predictions arising from this model. We show how thermal mixing of the stem cell population presents a barrier to clonal dominance through tracking of cell lineages. By abstracting the new model, we demonstrate how invariant fate progression in developing cells is achieved in a complex, multi-stable system. Finally, we use the model to explore how different physical mechanisms of cell signalling, division and apoptosis are compatible with known behaviours. 1834-Plat Phage DNA Dynamics in Correlation with Cell Fates Qiuyan Shao, Alexander Hawkins, Lanying Zeng. Biochemistry and Biophysics, Texas A&M University, College Station, TX, USA. Bacteriophage lambda begins its infection cycle by ejecting its DNA into its host E. coli cell, after which either the lytic or lysogenic pathway is chosen, resulting in different cell fates. In this study, using a new technique to monitor the spatiotemporal dynamics of the phage DNA in vivo, we found that the phage DNA moves via two distinct modes: localized motion and motion spanning the whole cell. One or the other motion is preferred depending on where the phage DNA is ejected into the cell. Through the phage DNA trajectories, we quantified the diffusion coefficient. Moreover, phage DNA motion is the same in the early phase of the infection cycle, irrespective of whether the lytic or lysogenic pathway is followed; hence, cell-fate decision-making appears not to be correlated with the phage DNA motion. However, after the cell commits to one pathway or the other, phage DNA movement slows during the late phase of the lytic cycle and remains the same during the entire lysogenic cycle. Throughout the infection cycle, phage DNA prefers the regions around the quarter positions of the cell. 1835-Plat Systems Mechano-Biology: Tension-Inhibited Protein Turnover is Sufficient to Physically Control Gene Circuits P.C. Dave P. Dingal, Dennis E. Discher. Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA, USA. Mechanotransduction pathways convert forces that stress and strain structures within cells into gene expression levels that impact development, homeostasis, and disease. The levels of some key structural proteins in the nucleus, cytoskeleton, or extracellular matrix have been recently reported to scale with tissue- and cell-level forces or mechanical properties, and so the mathematics of mechanotransduction becomes important to understand. Here, we show that if a given structural protein positively regulates its own gene expression, then stresses need only inhibit degradation of that protein in order to achieve stable, mechanosensitive gene expression. This basic ‘use it or lose it’ module is illustrated by application to meshworks of nuclear lamin A, mini-filaments of myosin II, and extracellular matrix collagen fibers - all of which possess filamentous coiled-coil/supercoiled structures. Past experiments not only suggest that tension suppresses protein degradation mediated and/or initiated by an enzyme, but also that transcript levels vary with protein levels as key transcription factors are regulated indirectly by these structural proteins. Coupling between modules 366a Tuesday, February 10, 2015 occurs within single cells and between cells in tissue, as illustrated during embryonic heart development where cardiac fibroblasts make collagen that cardiomyocytes contract. With few additional assumptions, the basic module has sufficient physics to control key structural genes in both development and disease. Platform: Ion Channel Regulatory Mechanisms 1836-Plat Coupling of Distinct Ion Channel Types in Neurons Mediated by AKAP79/ 150 Jie Zhang, Mark S. Shapiro. Physiology, UT Health Science Center, San Antonio, TX, USA. M-type Kþ channels, comprised of KCNQ2-5 (Kv7.2-7.5) subunits, play key roles in the regulation of neuronal excitability in the nervous system. In diverse neurons, L-type Ca2þ channels (LTCCs) drive transcriptional regulation via NFAT transcription factors, and in sensory neurons, TRPV1 cation channels excite neurons in response to heat, acidity or chemical ligands, driving nociception. The A-kinase-anchoring protein (AKAP)79/150 has been shown to orchestrate regulation of all three types of channels by PKC, PKA, calcineurin and NFATs. Using stochastic optical reconstruction microscopy (STORM) super-resolution microscopy, we have directly visualized individual signaling complexes containing AKAP79/150, these three ion channels and G proteincoupled receptors in neurons and tissue-culture cells. Using multi-color STORM, we observe AKAP150-mediated clustering of KCNQ, LTCCs and TRPV1 channels at the single-complex level. Thus, AKAP79/150 links different channel types together, raising the possibility of their functional, as well as physical, coupling. In sensory neurons, capsaicin caused PIP2 hydrolysis by TRPV1 activation. In neurons isolated from AKAP150þ/þ mice, brief application of low concentrations of capsaicin (100 nM), which we believe triggers only local PIP2 depletion, induced ~40% suppression of M-current (IM), suggesting close localization of TRPV1 and M-channels, the latter thus suppressed by TRPV1-induced local PIP2 depletion. However, in AKAP150-/neurons, IM was not affected by this modest activation of TRPV1 channels, implying the critical role of AKAP79/150. Application of the LTCC blocker, nifedipine, but not the N-type Ca2þ channel blocker, u-conotoxin GVIA, significantly suppressed desensitization and tachyphylaxis of TRPV1 currents, suggesting the functional coupling of LTCCs with TRPV1 channels, consistent with their physical coupling at the single-complex level seen with STORM. We thus find AKAP79/150 mediates physical and functional coupling of these three ion channels in sensory neurons, indicating physiological roles in tuning the nociceptive response to painful stimuli. 1837-Plat Stoichiometry of CRAC Channel Assembly and Gating Michelle Yen, Lumila A. Lokteva, Richard S. Lewis. Molecular and Cellular Physiology, Stanford University, Stanford, CA, USA. CRAC channels are opened by binding of the ER calcium sensor STIM1 to the C-terminus of the channel subunit Orai1. Previous functional experiments suggested a tetrameric channel stoichiometry, but the crystal structure of Drosophila Orai is a trimer of dimers, with each C-terminus forming a coiled-coil with its neighbor. This raises two fundamental questions: what is the stoichiometry of the CRAC channel, and does STIM1 bind to individual or pairs of C-termini to open it? To address these questions, we constructed hexameric concatemers of Orai1. Orai1 hexamers produced currents with properties that were indistinguishable from native ICRAC, including Ca2þ selectivity, Ca2þ-dependent inactivation, and modulation by 2-APB. The inhibitory effects of single L273D mutations confirmed that all 6 subunits participated equally in forming the functional channel. STIM1-Orai1 binding was studied using E-FRET between STIM1-YFP and CFP-Orai1. While the Orai1(L273D) C-terminus alone did not bind STIM1, it enhanced binding when paired with a neighboring WT C-terminus. To compare how monomer vs dimer binding are coupled to channel opening, we constructed hexamers with a single truncated or L273D C-terminus. The truncated hexamer showed significantly less activity than the L273D mutant, arguing against a pure monomeric gating mode. The relationship between STIM1 occupancy and channel activation was examined using Orai1 hexamers containing 1-3 STIM-binding mutations (producing 1-3 Orai1 heterodimers per channel). For both strong (L273D) and weak (L286S) inhibitory mutations, channel activity was well described by a model that assumes independent and equal energetic contributions from each heterodimer. In summary, we present the first functional evidence that hexameric Orai1 channels have the same properties as native CRAC channels. Our data suggest that STIM1 binds pairs of Orai C-termini and opens CRAC channels as a trimer of dimers, with each dimer contributing a constant amount of gating energy. 1838-Plat Structure and Selectivity in Bestrophin Ion Channels Tingting Yang1, Qun Liu2, Brian Kloss2, Renato Bruni2, Ravi C. Kalathur2, Youzhong Guo1, Edda Kloppmann3, Burkhard Rost3, Henry M. Colecraft1, Wayne A. Hendrickson1. 1 Columbia University, New York, NY, USA, 2New York Structural Biology Center, New York, NY, USA, 3TUM (Technische Universita¨t Mu¨nchen), Garching, Germany. Human bestrophin 1 (hBest1) is a calcium-activated chloride channel from the retinal pigment epithelium, where mutations are associated with vitelliform macular degeneration, or Best disease. We describe the structure of a bacterial homolog (KpBest) of hBest1 and functional characterizations of both channels. KpBest is a pentamer that forms a five-helix transmembrane pore, closed by three rings of conserved hydrophobic residues, and has a cytoplasmic cavern with a restricted exit. From electrophysiological analysis of structure-inspired mutations in KpBest and hBest1, we find a sensitive control of ion selectivity in the bestrophins, including reversal of anion/cation selectivity, and dramatic activation by mutations at the cytoplasmic exit. A homology model of hBest1 shows the locations of disease-causing mutations and suggests possible roles in regulation. 1839-Plat HCN Channels: The Molecular Basis for their cAMP-TRIP8b Regulation Andrea Saponaro1, Chiara Donadoni1, Sofia R. Pauleta2, Francesca Cantini3, Manolis Matzapetakis4, Gerhard Thiel5, Lucia Banci3, Bina Santoro6, Anna Moroni1. 1 Biosciences, University of Milan, Milan, Italy, 2REQUIMTE/CQFB, Department of Chemistry, New University of Lisbon, Lisbon, Portugal, 3 CERM, Department of Chemistry, University of Florence, Florence, Italy, 4 ITQB, New University of Lisbon, Lisbon, Portugal, 5Membrane Biophysics, Technical University of Darmstadt, Darmstadt, Germany, 6Department of Neuroscience, Columbia University, New York, NY, USA. Hyperpolarization-activated cyclic nucleotide-regulated (HCN1-4) channels are involved in the regulation of several higher order neural functions, such as short- and long-term memory processes (1). HCN channels are exquisitely sensitive to endogenous levels of cAMP, since they directly bind cAMP through a specialized domain in their cytoplasmic C-terminus (cyclic nucleotide binding domain, CNBD) (2). In addition to cAMP, HCN channels are further regulated by TRIP8b, their brain-specific auxiliary subunit. TRIP8b antagonizes the effect of cAMP on HCN channel opening, as it interacts with the CNBD of the channel (3). We employed solution NMR methodologies to determine the 3D structure of the human HCN2 CNBD in the cAMP-free form, and mapped on it the TRIP8b interaction site. Thus, we were able to reconstruct, for the first time, the molecular mechanisms underlying the dual regulation of HCN channel activity by cAMP-TRIP8b (4). Furthermore, site-directed mutagenesis followed by biochemical/biophysical analysis allowed us to identify key residues within the CNBD involved in TRIP8b binding. These new structural information will provide deeper insights into the molecular basis of neurological disorders associated with dysfunction of the HCN channel conductance in neurons, potentially leading to the design of drugs able to modulate HCN channel mediated memory processes. 1) Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsa´ki G, Siegelbaum SA, Kandel ER, Morozov A. (2004), Cell 119(5):719-732. 2) Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR. (2001), Nature 411(6839):805-10. 3) Hu L, Santoro B, Saponaro A, Liu H, Moroni A, Siegelbaum S. (2013), J Gen Physiol 142:599-612. 4) Saponaro A, Pauleta SR, Cantini F, Matzapetakis M, Hammann C, Donadoni C, Hu L, Thiel G, Banci L, Santoro B, Moroni A. (2014), PNAS Sep 2. pii: 201410389. [Epub ahead of print]. 1840-Plat Live Cell Biochemistry Implicates Protein Kinase a Modulation of L-Type CaV1.4 Channels Lingjie Sang, Ivy E. Dick, David T. Yue. Biomedical Engineering, Johns Hopkins University, Baltimore, MD, USA. The regulation of L-type Ca2þ channels by protein kinase A (PKA), though biologically crucial, has long remained mechanistically storied and complex, as studied in native cells at one extreme, and through in vitro biochemistry at the other. Here, we adopt a different tactic and focus initially on an intermediate context, using ideas drawn from synthetic biology and live-cell biochemistry. We set out to create a form of PKA modulation in L-type channels, based on our recent findings that: (a) calmodulin (CaM) competes for binding at a channel C-terminal ‘IQ’ domain with an ‘ICDI’ module in the C-terminal extremity Tuesday, February 10, 2015 of L-type CaV1.3/1.4 channels, and (b) dislodging CaM profoundly suppresses peak channel opening by severalfold and eliminates their Ca2þ-dependent inactivation (CDI) (Adams et al (2014), Cell in press). We reasoned that implanting a synthetic phosphorylation site in ICDI might weaken IQ interaction in a PKA-sensitive manner, allowing channels to rebind CaM and undergo CDI. Cognizant that full-bore PKA signaling is best conserved within certain native rather than model cells, we performed live-cell FRET interaction assays (IQ versus ICDI) in adult guinea-pig ventricular myocytes renown for strong PKA signaling. To our surprise in control experiments, we discovered that IQ interaction with the wild-type ICDI of L-type CaV1.4 channels is already sharply attenuated by PKA activation, whereas ICDI modules from other L-type isoforms showed no such modulability. Accordingly, we synthesized chimeric L-type CaV1.3 channels fused to the CaV1.4 ICDI module, and endowed such channels with robust forskolin-dependent enhancement of CDI, as observed in HEK293 cells. For wild-type CaV1.4 channels, we now also resolved analogous forskolin activatable CDI. This effect, discovered through a synthetic live-cell biochemical approach, might underlie the dopaminergic regulation of CaV1.4 implicated in circadian control within the retina. 1841-Plat A Comprehensive Search for Calcium Binding Sites Critical for TMEM16A Calcium-Activated Chloride Channel Activity Huanghe Yang, Jason Tien, Christian J. Peters, Xiu Ming Wong, Tong Cheng, Yuh Nung Jan, Lily Y. Jan. UCSF/HHMI, San Francisco, CA, USA. TMEM16A forms calcium-activated chloride channels (CaCCs) that regulate physiological processes such as the secretions of airway epithelia and exocrine glands, the contraction of smooth muscles, and the excitability of neurons. Notwithstanding intense interest in the mechanism behind TMEM16A-CaCC calcium-dependent gating, comprehensive surveys to identify and characterize potential calcium sensors of this channel are still lacking. By aligning distantly related calcium-activated ion channels in the TMEM16 family and conducting systematic mutagenesis of all conserved acidic residues thought to be exposed to the cytoplasm, we identify four acidic amino acids as putative calciumbinding residues. Alterations of the charge, polarity, and size of amino acid side chains at these sites alter the ability of different divalent cations to activate the channel. Our results thus demonstrate that direct binding of calcium to TMEM16A triggers channel activation independently of calmodulin, identify novel interaction sites between calcium ions and TMEM16A, and lay the groundwork for future studies examining the mechanism of calciumdependent TMEM16 channel activation. 1842-Plat Molecular Mechanism of Zinc Inhibition on Voltage-Gated Proton Channel Hv1 Feng Qiu1, Adam Chamberlin2, Sergei Noskov3, H. Peter Larsson1. 1 Physiology and Biophysics, University of Miami, Miami, FL, USA, 2 2Institute for BioComplexity and Informatics (IBI) and Department of Biological Sciences, University of Calgary, Calgary, AB, Canada, 3 University of Calgary, Calgary, AB, Canada. The voltage-gated proton channels (Hv1) have been shown to be involved in many physiological processes in which they play essential roles, such as pH homeostasis and charge compensation. The most potent inhibitor for Hv1 is zinc. For example: the quiescent sperm cells in the male reproductive system become active once introduced into the female reproductive tract by the removal of zinc inhibition. Zinc blocks the Hv1 current by shifting the voltage dependence of channel activation to a more depolarized range. In this study, we use voltage clamp fluorometry technique to identify the molecular mechanism of zinc inhibition on Hv1. We found that the zinc binding site is localized in each subunit of the dimeric Hv1 and that several polar amino acids on the extracellular part of this channel play different roles in the binding. Based on these results, we propose that there exist two sites for zinc occupancy: one is localized close to S1 and affects the voltage dependence of channel opening; the other one is localized in the proximity of the proton permeation pathway and thereby impairing the ion conduction of the channel. Since zinc is by far the only physiological extracellular blocker for Hv1, the detailed study of molecular mechanism of zinc binding will provide valuable information for future drug development for Hv1. 1843-Plat Alcohol Inhibition of a Chemically-Activated GIRK2 Channel Ian W. Glaaser1, Nidaa O. Marsh1, Senyon Choe2, Paul A. Slesinger1. 1 Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, USA, 2Structural Biology, Salk Institute, LaJolla, CA, USA. Alcohol is widely used and often abused. Yet, the molecular understanding of its action on brain function is poorly understood. Alcohol directly modulates 367a the activity of several ion channels, including the G protein-gated inwardlyrectifying potassium (GIRK) channel. GIRK channels are directly activated by alcohol, independent of their typical G-protein mediated pathway. Currently, however, the molecular mechanism underlying alcohol activation of GIRK is poorly understood. We recently demonstrated that introduction of a Cysteine into the alcohol pocket of GIRK2(L257C) created a channel that could be chemically activated with alcohol-like cysteine-reacting reagents. Here, we studied the channel gating properties of purified GIRK2L257C channels in a defined reconstituted system that allowed precise control of the lipids, G proteins and ions. We expressed a truncated cysless GIRK2-L257C(GIRK2D*-L257C) in Pichia pastoris, reconstituted purified protein into liposomes and studied the function of purified GIRK2D* L257C using a high throughput potassium flux assay. Reconstitution of GIRK2D*-L257C into POPE:POPG:PIP2 containing liposomes exhibited a basal, barium-sensitive flux that was potently enhanced by pre-incubation with MTS-hydroxyethyl(MTS-HE) as well as the Gbg G-protein subunits. Propanol treatment also enhanced the Kþ flux. Thus, in the absence of any other proteins or cytoplasmic regulators, these experiments demonstrate the direct activation of GIRK2 channels by three distinct ligands, Gbg G-proteins, alcohol and MTS-HE. Interestingly, MTS-HE-activated GIRK2D*L257C channels were inhibited by propanol in a dose-dependent manner. Inclusion of TCEP (tris(2-carboxyethyl)phosphine), which reduces disulfides, decreased MTS-HE activation but converted the propanol response from inhibition to activation. These experiments reveal that GIRK2 channels can also be inhibited by alcohol, perhaps through a different site, depending on the level of basal channel activation. Elucidating the details underlying alcohol’s effects on channel proteins is paramount to developing selective pharmacological tools that could be used in the treatment of alcohol abuse and addiction. Platform: Bioenergetics and Mitochondrial Signaling 1844-Plat Investigation of the Role of the Phospholipid Cardiolipin in Activating Respiratory Complex Activity Murugappan Sathappa1, Christine T. Schwall2, Matthew R. Greenwood1, Matthew G. Baile3, Steven M. Claypool4, Nathan N. Alder1. 1 Molecular and Cell Biology, University of Connecticut, Storrs, CT, USA, 2 Icahn School of Medicine, Mount Sinai Hospital, New York, NY, USA, 3 Physiology, Johns Hopkins University School of Medicine, Baltimore, MD, USA, 4Physiology, The Johns Hopkins University School of Medicine, Baltimore, MD, USA. Cardiolipin is an anionic phospholipid with a unique dimeric structure containing four fatty acids and two phosphate diesters. Within eukaryotic cells, cardiolipin resides predominantly in the energy-transducing mitochondrial inner membrane, where it mediates the assembly of respiratory chain supercomplexes, establishment of cristae morphology, and maintenance of membrane potential. Following its de novo synthesis in the inner membrane, nascent cardiolipin undergoes remodeling to produce a mature form of the lipid with largely unsaturated acyl chains. Abrogation of this remodeling cycle by dysfunction in the transacylase enzyme tafazzin underpins the heritable mitochondrial disorder Barth syndrome. Using an epistasis panel of yeast knockouts in the cardiolipin remodeling pathway, we have shown that remodeled and unremodeled cardiolpin support measurable features of oxidative phosphorylation to a similar extent, despite having marked differences in their acyl chain compositions. This has led us to analyze how physiochemical features other than acyl chain identity - namely the headgroup structure or the absence of a lipid tail - might explain the importance of cardiolipin remodeling. Using respiratory complex IV (cytochrome c oxidase) reconstituted into soluble nanoscale bilayers (nanodiscs) of defined lipid composition, we demonstrate the requirement of cardiolipin in activating respiratory complex redox activity. Moreover, performing fluorescence-based and electrokinetic measurements with model membrane systems, we have analyzed the interaction between divalent cations and cardiolipin-containing bilayers, as well as the proton dissociation behavior of cardiolipin variants. Using modeling that combines Gouy-Chapman-Stern formalism with Langmuir adsorption isotherms, our data indicate that cardiolipin variants bind divalent cations with similar affinities, but differ greatly with respect to cation-dependent alterations in headgroup packing in the bilayer interface. Moreover, our zeta potential measurements challenge the prevailing model that a bicyclic, resonance-stabilized headgroup structure maintains disparate pKa values of the two phosphate groups. 368a Tuesday, February 10, 2015 1845-Plat Bacterial Nanowires of Shewanella Oneidensis MR-1 are Outer Membrane and Periplasmic Extensions of the Extracellular Electron Transport Components Sahand Pirbadian1, Sarah E. Barchinger2, Kar Man Leung1, Hye Suk Byun1, Yamini Jangir1, Rachida A. Bouhenni3, Samantha B. Reed4, Margaret F. Romine4, Daad A. Saffarini3, Liang Shi4, Yuri A. Gorby5, John H. Golbeck2,6, Mohamed Y. El-Naggar1,7. 1 Physics and Astronomy, University of Southern California, Los Angeles, CA, USA, 2Biochemistry and Molecular Biology, Pennsylvania State University, University Park, PA, USA, 3Biological Sciences, University of Wisconsin, Milwaukee, WI, USA, 4Pacific Northwest National Laboratory, Richland, WA, USA, 5Civil and Environmental Engineering, Rensselaer Polytechnic Institute, Troy, NY, USA, 6Chemistry, Pennsylvania State University, University Park, PA, USA, 7Molecular and Computational Biology Section, Biological Sciences, University of Southern California, Los Angeles, CA, USA. Bacterial nanowires offer a pathway for extracellular electron transfer (EET) by linking the respiratory chain of bacteria to external surfaces, including oxidized metals in the environment and engineered electrodes in renewable energy devices. Specifically, nanowires of the model metal-reducing bacterium Shewanella oneidensis MR-1 were previously shown to be conductive under non-physiological conditions. Despite the global, environmental, and technological consequences of bacterial nanowire-mediated EET, the composition, electron transport mechanism, and physiological relevance of these appendages remain unclear. The nanowires of S. oneidensis MR-1 were previously thought, but never shown, to be bacterial pili. In addition, the transport mechanism through bacterial nanowires has been the subject of intense debate, with ‘‘metallic-like’’ band transport and multistep redox hopping between multiheme cytochromes as the two proposed mechanisms. Here we report the first in vivo observations of the formation and respiratory impact of nanowires in S. oneidensis MR-1. Using live fluorescence measurements and quantitative gene expression analysis, we demonstrate that S. oneidensis MR-1 nanowires are extensions of the outer membrane and periplasm, rather than pilin-based structures. We show, through immunolabeling, that multiheme cytochromes localize to nanowires, in turn supporting the multistep redox hopping model as the transport mechanism. Furthermore, these bacterial nanowires are associated with outer membrane vesicles, structures ubiquitous in Gram-negative bacteria, and occasionally appear as membrane vesicle chains that transition to smoother filaments. Redox-functionalized membrane and vesicular extensions may represent a general microbial strategy for electron transport and energy distribution. 1846-Plat Pigment-Specific Fluorescence Spectroscopy of Single Antenna Complexes in Solution Quan Wang, W.E. Moerner. Department of Chemistry, Stanford University, Stanford, CA, USA. Oligomerization plays a critical role in shaping the light-harvesting properties of many photosynthetic pigment-protein complexes (PPCs) in vivo, but a detailed understanding of this process at the level of individual pigments is still lacking. Here, aiming to study the effects of oligomerization in vitro, we designed a single-molecule approach to probe the emissive properties of individual pigment sites as a function of a PPC’s different oligomerization states. Our method, based on the principles of anti-Brownian electrokinetic trapping of single fluorescent molecules, step-wise photobleaching, and multi-parameter fluorescence spectroscopy, allowed pigment-specific spectroscopic information on single PPCs to be recorded in a non-perturbative aqueous environment with unprecedented detail. In particular, we focused on the monomer-to-trimer transformation of allophycocyanin (APC), which is the core antenna complex of cyanobacteria and red algae. At the monomer level, we found that the two phycocyanobilin (PCB) pigments (a and b) show similar but non-degenerate emission properties with asymmetric FRET. Interestingly, b acts as an emissive trap that quenches the emission from a on an intact monomer and can continue to act as a nonradiative trap for a after photo-damage. In the assembled trimers, two sets of emissive properties, one resembling the b site on the monomer and the other significantly red-shifted from either of the two sites on the monomer, were revealed at the single-pigment level. These observations suggest that the bathochromatic shift that accompanies trimer assembly of APC is localized on the a pigment site, which, in the pre-assembled (monomeric) state of the protein, is shielded and protected by the b pigment. 1847-Plat Automated Detection of Whole-Cell Mitochondrial Motility and its Dependence on Cytoarchitectural Integrity Judith Kandel1, Philip Chou1, David M. Eckmann2. 1 Bioengineering, University of Pennsylvania, Philadelphia, PA, USA, 2 Anesthesiology and Critical Care, University of Pennsylvania, Philadelphia, PA, USA. Current methodologies used for mitochondrial motility analysis tend to overlook either individual mitochondrial tracks or a cell-wide measure of motility. Furthermore, motility analysis of individual mitochondria is usually quantified by establishing an arbitrary threshold for ‘‘directed’’ motion. In this work, we created a custom, publicly available computational algorithm based on a previously published approach (Giedt et al, 2012) in order to characterize the distribution of mitochondrial movements at the whole-cell level, while still preserving information about individual mitochondria. The technique is easy to use, robust and computationally inexpensive. Images are first preprocessed for increased resolution, and then each mitochondrion is tracked based on object connectivity in space and time. When our method is applied to entire cells, we reveal that the mitochondrial net distances in fibroblasts follow a continuous lognormal distribution within a given cell or group of cells. This is in contrast to discrete categories used by others to bin mitochondria as either static or motile. Most mitochondria exhibit negligible movement, with the distribution center corresponding to a net distance of 132.3 nm. A few select mitochondria achieve net distances of several microns. The ability to model whole-cell mitochondrial motility as a lognormal distribution provides a new quantitative paradigm by which to compare mitochondrial motility in naı¨ve and treated cells. We further demonstrate that microtubule and microfilament depolymerization shift the lognormal distribution in directions which indicate decreased and increased mitochondrial movement, respectively. This shift occurs with distances of all magnitudes, even seemingly negligible ones. Our findings advance earlier work on neuronal axons (Morris and Hollenbeck, 1993) by relating them to a different cell type, applying them on a global scale, and automating measurement of mitochondrial motility in general. 1848-Plat Molecular Identity and Functional Characterization of Chloride Intracellular Channel (CLIC) Proteins in Cardiac Mitochondria Devasena Ponnalagu1, Jason Farber2, Sowmya Sukur3, Wenyu Xin3, Shubha Gururaja Rao1, Harpreet Singh1. 1 Pharmacology and Physiology, Drexel University college of medicine, Philadelphia, PA, USA, 2Bucknell University, Lewisburg, PA, USA, 3Drexel University, Philadelphia, PA, USA. CLIC proteins have six different paralogs in mammals (CLIC1-6) and exist in both soluble and integral membrane forms. Preconditioning rabbit ventriculocytes with IAA-94, an inhibitor of CLICs, before ischemia, increased myocardial infarction implicating CLICs role in cardioprotection. However, the molecular identity of cardiac CLIC, and the mechanism of CLICmediated cardioprotection is unknown. In this study, we focused on establishing the molecular identity, localization and functional role of CLICs in cardiac tissue of R. novergicus. Relative qPCR analyses indicate CLIC4, CLIC5 and CLIC1 as the most abundant CLICs present in the ventricle. CLIC4 (5050.1%) and CLIC5 (7553%) localize to the mitochondria of adult cardiomyocytes (n¼3). Cardiac CLIC4 and CLIC5 are present in 4052% and 7453% of Percoll-purified mitochondria (n¼3), respectively. Also, the only CLIC present in D. melanogaster showed mitochondrial localization in cardiac tubes. Further, IAA-94 partially blocked the channel activity of Percoll-purified mitochondrial membrane proteins reconstituted in a planar lipid bilayer. We also investigated the role of CLICs in modulation of ROS production by ETC. In the presence of succinate (complex II/III) and glutamate/malate (complex I), addition of 100 mM IAA-94 resulted in a robust release of ROS from mitochondria. The EC50 was found to be 16.5 þ: 0.15 mM (n¼3) for succinate. Initial fast release was followed by a significant reduction (82.553% n¼3, p<0.05) in rate of ROS production. The ROS release is specific to chloride conductance inhibition, as it also occurs with DIDS (ClC blocker). Blocking mPTP by cyclosporine A did not alter ROS release generated by CLIC inhibition indicating it is independent of mPTP (n¼3). In conclusion, we have deciphered the molecular identity of a cardiac CLIC, established its mitochondrial localization and a role of Chloride conductance in modulation of mitochondrial ROS generation. Tuesday, February 10, 2015 1849-Plat Mitochondrial NM23-H4/NDPk-D is Multifunctional: Fueling Mitochondrial GTPase OPA1 and Triggering Mitophagy Uwe Schlattner1,2, Mathieu Boissan3,4, Guillaume Montagnac3, Malgorzata Tokarska-Schlattner1,2, Ce´cile Cottet-Rousselle1,2, Ce´line Desbourdes1,2, Marie-Lise Lacombe4, Lorena Griparic5, Zhentai Huang6, Yulia Y. Tyurina6, Jian Fei Jiang6, Alexander M. van der Bliek5, Aure´lien Roux7, Philippe Chavrier3, Valerian E. Kagan6. 1 Joseph Fourier University - Grenoble 1, LBFA, Grenoble, France, 2Inserm, U1055, Grenoble, France, 3Institut Curie Research Center and CNRS UMR 144, Paris, France, 4Universite´ Pierre et Marie Curie, University Paris 06, and Saint-Antoine Research Center, INSERM UMR-S, 938, Paris, France, 5 Department of Biological Chemistry, David Geffen School of Medicine at University of California, Los Angeles, CA, USA, 6Center for Free Radical and Antioxidant Health, Department of Environmental and Occupational Health, University of Pittsburgh, Pittsburgh, PA, USA, 7Biochemistry Department, University of Geneva, & Swiss National Center for Competence in Research Program Chemical Biology, Geneva, Switzerland. NM23-H4/NDPK-D forms symmetrical homohexameric complexes in the mitochondrial inter-membrane space. The well-established function of NM23-H4 is phosphotransfer activity as a nucleoside diphosphate kinase, using mitochondrial ATP to regenerate NTPs, especially GTP. NM23-H4 also strongly binds in vitro to anionic phospholipids, mainly cardiolipin (CL), and in vivo to the mitochondrial inner membrane (MIM). Membranebinding seems to be important for close co-localization of NM23-H4 with mitochondrial OPA1, a dynamin-like GTPase, involved in fusion of MIM. NM23-H4/OPA1 association increases GTP-loading on OPA1. Like OPA1 loss-of-function, silencing of NM23-H4, but not cytosolic NM23-H1/H2, results in mitochondrial fragmentation, reflecting fusion defects. Thus, NM23-H4 interacts with and provides GTP to OPA1, similar to what is observed for cytosolic NM23 isoforms which interact with endocytic dynamin-1 and 2 and provide GTP for efficient dynamin-mediated endocytosis (Boissan et al.2014, Science 344:1510). Such close association allows these motor proteins to work with high thermodynamic efficiency. Earlier, we have shown that NM23-H4, when fully bound simultaneously to MIM and outer membrane (MOM), loses its kinase activity, but becomes competent to support intermembrane lipid transfer. This depends on the presence of the mitochondria-specific CL, and allows CL to move from its site of synthesis, MIM, to the opposed MOM (Schlattner et al.2013, JBC 288:111). Once CL is externalized at the mitochondrial surface, it can serve as a recognition signal for the autophageal machinery, leading to the elimination of damaged mitochondria. In cells treated with a protonophoric uncoupler, CCCP, CL externalization and mitophagy are stimulated only by transfection with NM23-H4 wild-type, but not R90D-mutant, incapable of CL binding. Similarly, in mouse lung epithelial cells, knocking-down NM23-H4 suppresses CL externalization and mitophagy. These findings suggest that NM23-H4 has dual functions in bioenergetics and lipid signaling leading to autophagy. Support: FRM,ARC,GEFLUC, NIH(U19AIO6802/HL114453), HFSP(RGP0013/2014). 1850-Plat VDAC Opening Drugs to Induce Mitochondrial Dysfunction and Cell Death Eduardo N. Maldonado1, Monika Gooz2, David N. DeHart1, John J. Lemasters3,4. 1 Drug Discovery & Pharmaceutical Sciences, Medical University of South Carolina, Charleston, SC, USA, 2Medicine, Medical University of South Carolina, Charleston, SC, USA, 3Drug Discovery & Pharmaceutical Sciences; Biochemistry & Molecular Biology, Medical University of South Carolina, Charleston, SC, USA, 4Institute of Theoretical and Experimental Biophysics, Pushchino, Russian Federation. Background: Mitochondrial membrane potential (DJ) and reactive oxygen species (ROS) formation depend on metabolite flux into mitochondria through voltage dependent anion channels (VDAC). Free tubulin closes VDAC, and high free tubulin levels decrease DJ in cancer cells. Erastin opens VDAC by antagonizing the inhibitory effect of tubulin. Here, we hypothesized that erastin and erastin-like compounds open VDAC, increase mitochondrial metabolism and ROS formation, and activate c-jun N-terminal kinase (JNK), leading to mitochondrial dysfunction and cell killing. Our AIM was to evaluate the effects of erastin/erastin-like compounds on DJ, ROS, NADH, JNK activation, cell killing and protection by antioxidants in HepG2 cells. 369a Methods: Confocal/multiphoton fluorescence microscopy assessed DJ (tetramethylrhodamine methylester), ROS (chloromethyldichlorofluorescein [cmDCF], MitoSOX Red) and NADH (autofluorescence). JNK was assessed by Western blotting and cell killing by propidium iodide assay. Results: Erastin increased DJ by 46% and NADH by 30% and blocked the depolarizing effect of microtubule destabilizers in HepG2 human hepatoma cells. Increased DJ after erastin/erastin-like compounds induced mitochondrial hyperpolarization that was followed by depolarization. Small molecules X1 and X2, identified in a high-throughput screening, caused a more rapid drop of DJ (<1 h) compared to erastin (3-4 h). Erastin, X1 and X2 also maximally increased cmDCF and MitoSOX Red fluorescence after 1-2 h. Additionally, JNK activation peaked at 60 min. JNK activation and ROS formation preceded mitochondrial depolarization. Cell killing promoted by X1 (93%) and X2 (76%) after 12 h was blocked by the antioxidant N-acetyl cysteine (100 mM). Conclusion: Mitochondrial hyperpolarization caused by VDAC opening drugs causes oxidative stress, which in turn leads to JNK activation, mitochondrial dysfunction and cell death that is prevented by antioxidants. Grants DK073336, DK037034 and 14.Z50.31.0028 to JJL and ACS 13-043-01 to ENM. 1851-Plat The 18kDa Translocator Protein Interacts with VDAC1 and Triggers a Ros-Mediated Inhibition of Mitochondrial Autophagy Michelangelo Campanella. 1 RVC and UCL Consortium for Mitochondrial Research, London, UK. The 18kDa Translocator Protein (herein TSPO) co-localises on the Outer Mitochondrial Membrane (OMM) with the Voltage Dependent Anion Channel 1 (VDAC1) and partakes in the transport of cholesterol. Overexpressed in several types of mammalian cancers it positively correlates with the aggressiveness of lesions as well as inflammation of the brain therefore exploited as biomarker and target for suitable therapies. In Mouse Embryonic Fibroblasts (MEFs), the recombinant TSPO overexpression prevents Parkin mediated ubiquitination of mitochondrial proteins and recruitment of p62/SQTSM1 and LC3, thus leading to accumulation of dysfunctional mitochondria. Live cell imaging approaches demonstrate that TSPO enriched, mitophagy-evading, mitochondria present: i) defective Ca2þ signalling, ii) reduced coupling, iii) low ATP synthesis and iv) aberrant network morphology. The inhibition of mitochondrial ubiquitination by TSPO is independent from cholesterol trafficking and consequent of the overproduction of Reactive Oxygen Species (ROS) caused by defective mitochondrial metabolism. The prevention of ubiquitination likewise the suppression of cellular mitophagy are lost in MEFs knocked out for VDAC1 (VDAC1-/-) with which TSPO interacts to impair mitochondrial signalling and trigger oxidative stress. These data proposes TSPO as a novel regulatory element of the mitochondrial quality control and a further molecular determinant for the Parkin-mediated ubiquitination and removal of disposable mitochondria. Workshop: Managing Data and Statistics in the Informatics Era 1852-Wkshp Informatics Approaches to Data Preservation and Analysis in Protein Electrostatics Nathan A. Baker1, Chase Dowling1, Luke Gosink1, Trenton Pulsipher1, Susanna-Assunta Sansone2. 1 Pacific Northwest National Laboratory, Richland, WA, USA, 2Oxford eResearch Centre, Oxford, United Kingdom. Scientific data curation has presented a challenge to multiple disciplines in terms of preserving data, supporting its reproducibility, and enabling ready access of the data for future statistical analysis. We have addressed this challenge in the context of experimental and computational protein titration data. In particular, we have leveraged the ISA-TAB community-supported datasharing standard (http://www.isa-tools.org/) to collect and preserve protein pKa data. This data was collected from a variety of published and unpublished sources associated with pKa Cooperative (http://pkacoop.org), a group of researchers dedicated to advancing the understanding of protein electrostatics. Additionally, we have demonstrated the utility of collecting data in a standard format such as ISA-TAB by developing a new statistical pKa prediction approach which combines computational results from the pKa Cooperative effort into an aggregate classifier with significantly improved predictive power. 370a Tuesday, February 10, 2015 1853-Wkshp A Physicist’s Approach to Statistical Analyses of Biological Data Patrice Koehl. Computer Science and Genome Center, University of California, Davis, Davis, CA, USA. The ongoing transformation of biology to a quantitative discipline has drastically increased our opportunities to unravel the mechanisms that relate the dynamics of biological systems to their functions as it allows for the investigation of such systems at spatial and temporal scales never observed before. The biggest challenge today is to assimilate the wealth of information generated in this process into a conceptual framework. We face issues with the volume of data generated (a Big Data challenge) as well as with the complexity of the systems they represent. In this talk I will show examples for which a combination of mathematics, physics, and biology provides solutions to these challenges. I will focus specifically on the concept of networks in biology, their morphologies and dynamic behaviors. 1854-Wkshp Glycan Biosynthesis: Structure, Information, and Heterogeneity Anjali Jaiman, Mukund Thattai. Simons Centre for the Study of Living Machines, National Centre for Biological Sciences, Bangalore, India. The surfaces of all living cells are decorated with branched sugar polymers known as glycans. These information-rich structures confer cells with a recognizable molecular identity, and underlie many specific cell-cell interactions. Analytic methods - including NMR, mass-spectrometry, and glycan arrays - now permit the routine profiling of glycans associated with various cells or proteins. This has stimulated efforts to build comprehensive and searchable glycan databases. However, from an informatics perspective glycans present multiple challenges. First, whereas nucleotide and amino acid chains are efficiently represented as strings, sugars can polymerize into complex tree-like objects. The potential combinatorial space of glycans is therefore much larger than that of proteins. Second, many specific molecular interactions appear to be mediated by groups of closely-related glycan variants rather than by a single well-defined structure. This phenomenon of ‘‘micro-heterogeneity’’ makes it difficult to rigorously characterize the glycan repertoire of a cell. In this workshop, I will use ideas from algorithmic self-assembly to show that glycan structure and diversity are best understood through the lens of glycan biosynthesis. I will demonstrate that a specific glycan structure is the outcome of glycosyltransferase enzymes acting according to simple rules in a specific order, like workers on a factory floor. Errors in this process produce a well-defined spectrum of glycan by-products, precisely matching the observed micro-heterogeneity in real glycan profiles. This predictive theoretical framework allows us to use glycans as sensitive cell-biological probes. It provides a unifying perspective within which the rich and growing datasets of glycan structures can be organized and fully utilized. 1855-Wkshp Large-Scale Machine Learning Approaches for Molecular Biophysics Arvind Ramanathan1, Chakra S. Chennubhotla2, Pratul K. Agarwal3, Christopher B. Stanley4. 1 Computer Science, Oak Ridge National Lab, Oak Ridge, TN, USA, 2 Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, PA, USA, 3Computer Science and Mathematics Division, Oak Ridge National Lab, Oak Ridge, TN, USA, 4Neutron sciences Division, Oak Ridge National Lab, Oak Ridge, TN, USA. Extracting knowledge from large, heterogeneous, unstructured and highdimensional data is one of the major challenges for large-scale machine learning algorithms. In this talk, I will present our recent results developing unsupervised machine learning approaches to explore such data sets. A large number of these datasets follow heavy-tailed distributions, characterized by long-range dependencies. We quantify the tails of these distributions using higher order statistics and use tensor-based representations to build data mining algorithms for: (1) online detection of events that signify anomalies in spatio-temporal patterns; (2) building low- dimensional latent variable models to capture the intrinsic multiscale structure; and (3) hierarchical clustering and visual organization of the data to gain relevant insights. We will illustrate these approaches on a variety of applications including the integration of sparse experimental observations with atomistic-scale information for understanding the function of cellular systems. We will also discuss how these approaches can be widely applied to other domains. Workshop: Advances in Computing Large Systems 1856-Wkshp Reversible Folding of Hyperstable RNA Tetraloops Using Molecular Dynamics Simulations Angel E. Garcia1, Jacob Miner2, Alan A. Chen3. 1 Dept Phys/Appl Phys/Aston, Rensselaer Polytechnic Inst, Troy, NY, USA, 2 Dept of Biology, Rensselaer Polytechnic Inst, Troy, NY, USA, 3Dept of Chemistry, SUNY Albany, Albany, NY, USA. Structured RNAs exhibit a distinct preference for loops of precisely 4 nucleotides. Approximately 70% of these ‘‘tetraloops’’ are comprised of just three specific loop sequences: UUCG, GCAA, or CUUG. The abundance of these sequences is thermodynamic in origin, as each motif forms a unique network of non-canonical interactions within their loops that stabilize the folded state. Modification to the Amber force field enables the de-novo folding of three hy˚ RMSD from their experimentally deterperstable RNA tetraloops to 1-3 A mined structures using molecular dynamics simulations initialized in the unfolded state. To study the thermodynamics and kinetics of folding on an RNA tetraloop we simulated the (rGCAA) tetraloop with stem lengths of two (octamer), and four (dodecamer) C-G base pairs. The thermodynamics is obtained from replica exchange molecular dynamics (REMD) simulations with overall sampling exceeding 300 microseconds. The kinetics of the octamer was studied in a 100 microseconds molecular dynamics simulation using the Anton supercomputer. The thermodynamics reveal that the octamer folds and unfolds reversibly. However, the dodecamer behaves glassy and adopts multiple metastable loop configurations that do not unfold in extensive REMD simulations. The ability to recapitulate the signature non-canonical interactions of the three most abundant hyperstable stem-loop motifs represents a significant step towards the accurate description of nucleic acid tertiary structures, dynamics and stability using unbiased all-atom molecular dynamics simulations. 1857-Wkshp Bacterial Outer Membranes and Interactions with Membrane Proteins Wonpil Im. Department of Molecular Biosciences and Center for Bioinformatics, The University of Kansas, Lawrence, KS, USA. Because the bacteria’s outer membrane (OM) acts as an effective barrier against the permeation of both hydrophobic and hydrophilic compounds, gram-negative cell permeation is one of the biggest challenges to the discovery of novel antibiotics for bacterial infections and antibiotic resistance worldwide. The bacterial OM is a unique and highly asymmetric lipid bilayer composed of phospholipids in the inner leaflet and lipopolysaccharide (LPS) in the outer leaflet. An LPS molecule is a complex amphiphatic compound consisting of lipid A, a core oligosaccharide, and an O-antigen polysaccharide. Despite the direct relationship of gram-negative bacteria to the public health and also the fact that there are over 190 identified O-serotypes for Escherichia coli, our molecular-level understanding of how the bacterial OMs behave and work for various types of bacteria, how membrane proteins behave in the OM, and how known drug molecules and potential drugs can enter through the OMs is rudimentary at best. This talk presents our ongoing efforts on all-atom modeling and simulations of these complex bacterial OMs with and without various outer membrane proteins using the CHARMM36 (protein, lipid, carbohydrate) force fields. In addition, various technical aspects and perspectives are also discussed together with future developments of CHARMM-GUI LPS Modeler and OM Builder. 1858-Wkshp Protein Folding and Recognition in the Cell – an in Silico Approach Margaret S. Cheung1,2. 1 University of Houston, Houston, TX, USA, 2Rice University, Center for Theoretical Biological Physics, Houston, TX, USA. I will review the approach of coarse-grained molecular simulations for the investigation of protein folding and protein-protein interactions in a cellular environment, particularly the one for the research from my group. We used a low-resolution model for the representation of proteins and macromolecules that mimic a jam-packed space inside a cell. We made these low-resolution models act like ‘‘a real thing’’ by keeping the physics in its dynamics and the principle of chemical interactions between macromolecules in the simulations. With this approach, we were able to characterize the mechanism of protein folding and protein-protein interactions that involve structurally large rearrangement in the presence of dominant forces inside a cell, such as the volume exclusion from the macromolecular crowding effect and the ionic strength. Based on simple ideas for modeling a large system, I will report several new discoveries and testable predictions from our computational studies. Tuesday, February 10, 2015 1859-Wkshp Advances in Atomic-Level Simulations of Large-Scale Functional Motions of Membrane Transporters Emad Tajkhorshid, Mahmoud Moradi, Jing Li, Po-Chao Wen, Sundar Thangapandian, Josh Vermaas. Department of Biochemistry, and Beckman Institute, University of Illinois, Urbana Champaign, Urbana, IL, USA. Membrane transporters are specialized molecular devices that use various forms of cellular energy to drive active transport of their specific substrates across the membrane. Their fundamental role in diverse key biological processes has placed them among central drug targets, furthering widespread interest in their biophysical and mechanistic studies at a molecular level. Large-scale conformational changes are central to the function of membrane transporters. Description of these structural changes, however, requires sampling high-dimensional free energy landscapes that are inaccessible to conventional sampling techniques such as regular molecular dynamics (MD) simulations. We have recently developed a novel computational approach that, while being numerically expensive, has been the most efficient way to describe large-scale structural transitions in membrane transporters (as well as for any other macromolecular systems) using nonequilibrium methods employing system-specific collective variables, and a novel combination of several state-of-the-art sampling techniques [Moradi and Tajkhorshid, PNAS 110:18916-21 (2013); JCTC 10: 2866-80, 2014]. The approach is based on loosely coupled, multiple-copy MD simulations of large macromolecular systems preserving realistic representations of the systems in explicit membranes, and therefore relies on massive computing resources. Here we describe the application of the methodology to the study of several classes of membrane transporters, in order to characterize the inter-conversion of these molecular devices between the major conformational states necessary for their function, to characterize the free energy profiles associated with these transitions, and more importantly how chemical details such as ion/substrate binding drastically modulate the energy landscapes. The results of these simulations elucidate highly relevant mechanistic details of the function of membrane transporters providing a detailed structural basis for the experimentally observed phenomena. Workshop: Microfluidics Tools for Studying Molecules and Cells 1860-Wkshp Integrated Microfluidic Devices for Studying Aging and Adhesion of Individual Bacteria Stephen C. Jacobson1, Joshua D. Baker1, David T. Kysela2, Yves V. Brun2. 1 Department of Chemsitry, Indiana University, Bloomington, IN, USA, 2 Department of Biology, Indiana University, Bloomington, IN, USA. Analysis of single cells provides powerful insight into biological processes that are often missed when a population of cells is studied as an ensemble. We are developing microfluidic-based approaches coupled with optical microscopy to track individual bacteria and to improve the temporal and spatial resolution of single-cell measurements. The microfluidic devices automate the steps of cell culture, synchronization, reagent delivery, and analysis. To study cell growth and aging, we integrated nanochannel arrays into the microfluidic devices that physically trap bacteria. The bacteria grow and divide along the nanochannels in one-dimension, and parent cells and their progeny are easily tracked from generation to generation. With these devices, we are able to determine cell growth and division rates, accumulation of cellular damage over time, and inheritance from one generation to the next. For the adhesion studies, swarmer cells are synchronized in our ‘‘baby machine’’ and delivered to adjacent microchannels where we monitor adhesion of individual bacteria to channel surfaces. To better understand how bacteria attach to surfaces, rates of attachment of mutant strains that lack pili and motility are compared to the rate of attachment of wild-type cells. 1861-Wkshp Democratization of Next-Generation Imaging, Diagnostics and Measurement Tools through Computational Photonics Aydogan Ozcan. Electrical Engineering & Bioengineering, University of California, Los Angeles, Los Angeles, CA, USA. My research focuses on the use of computation/algorithms to create new optical microscopy, sensing, and diagnostic techniques, significantly improving existing tools for probing micro- and nano-objects while also simplifying the designs of these analysis tools. In this presentation, I will introduce a new set of computational microscopes which use lens-free on-chip imaging to replace traditional lenses with holographic reconstruction algorithms. Basically, 3D images of specimens are reconstructed from their ‘‘shadows’’ providing considerably improved field-of-view (FOV) and depth-of-field, thus enabling large 371a sample volumes to be rapidly imaged, even at nanoscale. These new computational microscopes routinely generate >1-2 billion pixels (giga-pixels), where even single viruses can be detected with a FOV that is >100 fold wider than other techniques. At the heart of this leapfrog performance lie self-assembled liquid nano-lenses that are computationally imaged on a chip. The field-ofview of these computational microscopes is equal to the active-area of the sensor-array, easily reaching, for example, >20 mmt2 or >10 cmt2 by employing state-of-the-art CMOS or CCD imaging chips, respectively. In addition to this remarkable increase in throughput, another major benefit of this technology is that it lends itself to field-portable and cost-effective designs which easily integrate with smartphones to conduct giga-pixel tele-pathology and microscopy even in resource-poor and remote settings where traditional techniques are difficult to implement and sustain, thus opening the door to various telemedicine applications in global health. Some other examples of these smartphonebased biomedical tools that I will describe include imaging flow cytometers, immunochromatographic diagnostic test readers, bacteria/pathogen sensors, blood analyzers for complete blood count, and allergen detectors. Through the development of similar computational imagers, I will also report the discovery of new 3D swimming patterns observed in human and animal sperm. 1862-Wkshp A Microfluidic Rapid Freeze Quench Apparatus for High Field EPR Measurements Alberto Collauto, Royi Kaufmann, Daniella Goldfarb. Chemical Physics, Weizmann Institute of Science, Rehovot, Israel. Rapid freeze quench (RFQ) EPR is a well-established technique for trapping reaction intermediates. A major difficulty in using commercial RFQ-EPR combined with standard X-band EPR spectroscopy is the relatively large amount of sample needed for each time point, and the associated amount that is wasted in the dead volume of the tubes and mixer. This is particularly prohibitive when one would like to couple RFQ with high resolution EPR techniques such as ENDOR (electron-nuclear double resonance) and DEER (electron-electron double resonance) that provide electron-nuclear and electron-electron distances and are less sensitive than standard continuous wave EPR. We have developed a dedicated microfluidic RFQ (RFQ) apparatus for W-band measurements, optimized for the small W-band sample size and a minimal sample amount for a series of ~7 time points collected in triplicates (~200 l of 0.03-0.1 mM labeled protein). The mixer is based on a recent published design[1] with a modified sample ejection system and cold trap. It current time window is 5-90 msec and its performance has been demonstrated on the reduction of nitroxide with dithionite[2]. The current state of the RFQ apparatus is demonstrated on : (i) the ATPase activity of an RNA helicase. Here Mg2þ was substituted with Mn2þ and the 31P ENDOR spectrum was recorded. The ADP and ATP spectra have significantly different lineshapes and can therefore be used to probe the hydrolysis state. (ii) Conformational changes induced in a protein through ligand binding as detected by DEER. 1. Egawa T, Durand JL, Hayden EY, Rousseau DL, Yeh S-R. Anal Chem. 2009;81(4):1622-7. 2. R. Kaufmann, D. Goldfarb.. J Magn Reson. 2013;230:220-6. 1863-Wkshp Cell and Vesicle Analysis in Microchambers Petra S. Dittrich. Biosystems Science and Engineering, ETH Zurich, Zurich, Switzerland. Microfluidics is nowadays an established technology and provides as a huge toolbox for analytical and bioanalytical methods. Microfluidic platforms facilitate precise handling and positioning of cells, creating of chemically defined liquid environments, and tailoring mechanical or physical conditions. In recent years, we have developed several microfluidic platforms for single cell analysis. Combinations of cell trapping and encapsulation in microchambers accommodating volumes of tens to hundreds of picoliters facilitated the analyses of living cells and the chemical analysis of cell lysates. Moreover, we integrated these techniques with immunological methods, which allowed the quantification of proteins and other biomolecules with unprecedented high sensitivity. The microfluidic platforms proved highly useful for analysis of giant unilamellar vesicles (GUVs), which we create in order to elucidate processes at the membrane. We could address questions of membrane permeability and membrane fusion. In addition, we could gain new insights into the properties of membranes, when exposed to mechanical forces. More precisely, we used vesicles with phaseseparated domains and deformed them while they are trapped in the microfluidic device. We use confocal laser scanning microscopy to image the GUVs and could show that lipid sorting occurs, i.e., domains fuse, upon the increase of tension. Occasionally, we even observe budding of the liquid disordered phase. In further experiments, we exposed the GUVs to shear stress of defined strength and were able to visualize the changes of the domain shape and their relaxation after stopping the shear stress. Together, these studies may reveal in more detail the role of the membrane in the cellular response to mechanical strains. 372a Tuesday, February 10, 2014 Posters Protein Structure and Conformation III 1864-Pos Board B1 The Mechanoenzymatic Properties of Drp1 in Nucleotide Induced Constriction of Lipid Bilayers Christopher A. Francy, Frances J.D. Alvarez, Louie Zhou, Jason A. Mears. Pharmacology, Case Western Reserve University, Cleveland, OH, USA. Mitochondria are dynamic organelles that continually undergo cycles of fission and fusion. Dynamin-related protein 1 (Drp1), an 81 kDa GTPase, is the main mediator of mitochondrial fission. In order to mediate fission, Drp1 is thought to form large oligomers in the presence of nucleotide on the outer mitochondrial membrane. Using sedimentation assays and electron microscopy, we confirm that Drp1 selfassembles in the presence of either or both nucleotide and artificial lipid bilayers in vitro. We further identify these oligomeric species as conformationally distinct structures. Drp1 constricts lipid bilayers through a mechanism that requires GTP hydrolysis. Following constriction, Drp1 disassembles and can reassociate with lipid bilayers. Here we also show that the variable domain (VD) is not required for lipid association. Rather, it limits Drp1 oligomerization and aids in oligomer curvature. Our results support the conclusion that Drp1 is a mechanoenzymatic protein regulated through distinct interactions with nucleotide and liposomes. 1865-Pos Board B2 Caveolin Revealed: A Mutagenesis Study of Caveolin-1 Sarah Plucinsky, Kerney J. Glover. Lehigh University, Bethlehem, PA, USA. Caveolae are 50-100 nm invaginations in the plasma membrane that are rich in cholesterol, sphingomyelin and the integral membrane protein called caveolin1. Caveolin-1 has three main functions: forming caveolae, cell signaling, and endocytosis. To examine the importance of conserved residues within the scaffolding and intra-membrane domains of caveolin-1, alanine and phenylalanine scanning mutagenesis was performed on conserved residues. These residues were identified by sequence alignment of the three caveolin isoforms. Sixteen residues (S88, F92, K96, Y97, Y100, L103, P110, A112, G116, F119, A120, S123, H126, I127, W128 and P132) were mutated individually to both alanine and phenylalanine and subjected to analysis by NMR spectroscopy (1H-15NHSQC). The effect of the mutation on the overall protein structure was monitored by comparison to the wild-type spectrum. These mutagenesis studies reveal seven residues (Y100, P110, A112, G116, S123, H126, and P132) that are structurally significant. The residues within the scaffolding domain are more permissive to mutation than the intra-membrane domain. The proline residues within the intra-membrane domain were shown to be critical for protein structure. These findings shed light onto the structurally relevant residues within the caveolin-1 scaffolding and intra-membrane domains, and further our understanding of the requirements for protein structure and stability. 1866-Pos Board B3 Reconstitution and Topological Analysis of Caveolin-1 in Bicelles Kyle Root. Chemistry, Lehigh University, Bethlehem, PA, USA. Caveolae are 50-100 nm invaginations in the plasma membrane of many cell types that play a critical role in signal transduction. Caveolin-1 is a 21 kD integral membrane protein that is required for the formation of caveolae. Caveolin-1 is thought to adopt an unusual horseshoe topology in the membrane where the Nand C- termini are cytoplasmic. Caveolin-1 has 4 native tryptophan residues in this region (W85, W98, W115, and W128) that can be used as reporters of the micro-environment of the polypeptide chain. Caveolin-1 single tryptophan mutants were successfully reconstituted into CHAPSO/DMPC bicelles. Fluorescence emission measurements were performed on each mutant and analysis of these spectra indicated that tryptophan residues 85 and 128 are in the head group region of the lipid bilayer with lmax values of 344.4 5 2.4 nm and 338.2 5 0.6 nm respectively, and tryptophan residues 98 and 115 are inserted into the hydrophobic core with lmax values of 334.4 5 0.2 nm and 330.2 5 1.0 nm, respectively. The information gleaned from these studies was supported by MD simulations performed on caveolin-1 in a DMPC bilayer. These data together support the postulation that caveolin-1 contains a membrane embedded turn. 1867-Pos Board B4 Fragment-Based Drug Design Approach for Targeting Phospholipid Biosynthesis Pathway in Plasmodium Falciparum Ewelina Guca1, Marina Lavigne1, Franc¸ois Hoh2, Jean-Franc¸ois Guichou2, Christian Roumestand2, Henri Vial1, Rachel Cerdan1. 1 DIMNP UMR 5235, University of Montpellier II, Montpellier, France, Centre de Biochimie Structurale, INSERM/CNRS UMR 5048, Montpellier, France. Phospholipid synthesis metabolic pathways in Plasmodium falciparum are validated drug targets for new type of antimalarials. In the de novo Kennedy pathway of phosphatidylcholine biosynthesis, the second step catalyzed by CTP:phosphocholine cytidylytransferases [2.7.7.15] is rate limiting and appears essential for the parasite survival at its blood stage. We are focused on the structural characterization of this enzyme, the identification of effectors by fragment-based drug design approach (FBDD) and then their optimization to eventually design a lead. We solved the first reported crystal structure of ˚ and the catalytic domain of the enzyme target (PfCCT) at resolution 2.2 A ˚ that the enzyme-product (CDP-choline) complex structure at resolution 2.4 A give detailed images of binding pocket and demonstrate conformational changes between apo- and holo-protein forms at atomic level. The FBDD method uses a library of small molecules (fragments) with molecular weight that does not exceed 300 Da to explore target binding sites. Primary screening of fragment library (230 molecules) has been investigated by fluorescencebased thermal shift assay and Nuclear Magnetic Resonance Saturation Transfer Difference (NMR STD) method is used as a secondary screen to eliminate false positive ligands. This combination of techniques identified so far 4 fragment hits that are currently evaluated for their binding modes and affinities. Cocrystallization of the protein-fragments complexes is carrying out to provide accurate information on the binding modes of the small molecules and topology of interactions will be used to rationally monitor every iterative round of the optimization process allowing subsequent rational design. 2 1868-Pos Board B5 Mechanisms of Pin1 Regulation of IRAKM Stability in Toll-Like Receptor/Interleukin-1 Receptor Signaling Jeahoo Kwon1, Morris Nechama2, Kun Ping Lu2, Linda K. Nicholson1. 1 Molecular Biology and Genetics, Cornell University, Ithaca, NY, USA, 2 Harvard Medical Center, Harvard Univeristy, Boston, MA, USA. The molecular mechanisms of asthma, a chronic inflammatory sickness that involves activation of the innate immunity signaling pathways, are not yet fully understood. Our studies focus on the peptidyl-prolyl isomerase enzyme Pin1 and its substrates in innate immunity signaling. Prolyl peptide bonds, such as in pS/T-P motifs, can exist in two distinct isomer conformations, cis and trans, that exchange on a slow time scale (several minutes). Isomerization of pS/T-P motifs can be accelerated by Pin1. Pin1 has recently been shown to be activated by IL-33 which activates IL-1R signaling in Th2 cells, inducing asthma. Pin1 knockout (KO -/-) abolishes Th2 cytokine release thereby preventing asthma symptoms. Pin1 also prevents degradation of IRAKM, a negative regulator of TLR/IL-1R signaling, but promotes the nuclear localization of IRAKM. IRAKM is phosphorylated upon IL-33 treatment. The phosphomimetic mutation IRAKM S110E is dramatically stabilized in a Pin1-dependent manner, even more than wild type IRAKM. We test the model that Pin1 regulation of IRAKM stability and nuclear localization is mediated by the direct interaction of Pin1 with phosphorylated IRAKM. The dissociation constants, KD, for Pin1 binding to phosphorylated IRAK-M (pIRAKM) and to the phosphomimetic mutation IRAKM S110E were determined using the 1H 15N HSQC NMR experiment. Furthermore, the Pin1 catalyzed isomerization of wild type pIRAKM and IRAKM S110E were measured using the ROESY NMR experiment. This study is expected to contribute to our understanding of the novel mechanism of Pin1 regulation of the TLR/IL-1R signaling pathway in terms of specific domains of IRAKM. 1869-Pos Board B6 Using Biochemical and Structural Approaches to Study ErbB2-Containing Heterodimers Lily L. Raines, Daniel J. Leahy. Biophysics and Biophysical Chemistry, Johns Hopkins University School of Medicine, Baltimore, MD, USA. Dimerization of epidermal growth factor receptor (EGFR/ErbB) homologs is necessary for their activation. Dysregulation of each of these receptors is linked to a variety of cancers. Most members of this protein family adopt a tethered conformation in the absence of ligand that prevents the formation of active dimers, while ErbB2 adopts a constitutively extended conformation mimicking the ligand-bound conformation. Surprisingly, ErbB2 does not homodimerize yet is the preferred dimerization partner of the other EGFR homologs. The reasons for this are unclear, and previous attempts to address these questions using structural techniques were unsuccessful. Here we drove dimerization of the extracellular domains of EGFR family members with ErbB2 by fusion of one extracellular domain to the light and the other to the heavy chain of an Tuesday, February 10, 2014 antibody. We pursued crystallization of these heterodimers, and currently use small angle x-ray scattering and electron microscopy to determine the structure of these heterodimers in the presence and absence of ligand to reveal what structural facets underlie their activation. 1870-Pos Board B7 Probing the Cellular Entry Pathway via TolC of the Cytotoxin, Colicin E1 Karen S. Jakes1, Stanislav D. Zakharov2, Xin S. Wang2, Ilya Seleznev2, William A. Cramer2. 1 Physiology & Biophysics, Albert Einstein College of Medicine, Bronx, NY, USA, 2Department of Biological Sciences, Purdue University, W. Lafayette, IN, USA. The pathway of cellular entry of colicins and viral DNA is a fundamental structure problem that is relevant to an understanding the molecular basis of infectious diseases. The cytotoxin colicin E1 uses the outer membrane/transperiplasmic drug export protein, TolC, for its import. TolC consists of a 12 strand OM b-barrel connected to a 12 strand a-helical tunnel that defines a pathway through the peptidoglycan barrier in the periplasm to the cytoplasmic membrane in which the C-terminal domain of colicin E1 inserts and forms a depolarizing ion channel. The nature of the interaction of the colicin with TolC and the mechanism of its translocation are unknown. Previous studies with planar bilayers showed that colicin E1 occludes TolC channels [1], as do certain colicin T-domain peptides [2]. Here, in vivo protection of sensitive E. coli from colicin E1 by a series of N-terminal colicin peptides is used to probe the interaction of the colicin with TolC, with the goal of defining the sites of TolC-colicin interaction and the mechanism of colicin entry. N-terminal segments ‘1-40’,’1-81’, and ‘1-100’ of the colicin did not provide cytotoxic protection, nor occlude TolC channels. Segments ‘1-120’, ‘1-140’, 1-190’, as well as ‘41-190’ and ‘57-190’ protected efficiently in vivo and occluded TolC with high efficiency. Occlusion required a trans-negative electrical potential and was irreversible. Co-elution of the colicin peptides with TolC on a Superdex 200 column was shown for ‘41-190’, but not for ‘1-81.’ In addition to the correlation with protection in vivo from killing by colicin E1, occlusion efficiency also correlated with a basic pI between colicin residues 82 and 140. [1] Biophys. J., 87:39013911, 2004; [2] Biochem. Soc. Trans., 40:1463-1468, 2012. Support: NIH AI091633 (KSJ); NIH GM38323 and the Henry Koffler Professorship (WAC). 1871-Pos Board B8 The Membrane Catalysis Model: Apelin and its Receptor Robin E. Patterson1, Nathan Weatherbee-Martin1, Nigel A. Chapman1, Denis J. Dupre´2, Jan K. Rainey1,3. 1 Department of Biochemistry & Molecular Biology, Dalhousie University, Halifax, NS, Canada, 2Department of Pharmacology, Dalhousie University, Halifax, NS, Canada, 3Department of Chemistry, Dalhousie University, Halifax, NS, Canada. Apelin is a peptide hormone that activates the class A G-protein coupled apelin receptor. Apelin is found in several bioactive isoforms in the body, ranging from 12 to 55 amino acids in length. Apelin-17 has previously been shown to bind to micelles of anionic detergents, which implies that apelin may interact with cell membranes prior to receptor activation. It has been theorized that this interaction with the membrane may induce conformational changes necessary for peptide recognition by the receptor, while increasing local concentration of the ligand in what is known as the membrane catalysis hypothesis. Here, we describe a method for conjugating various isoforms of apelin to fluorophores, while still maintaining bioactivity, as demonstrated by a phosphorylated extracellular signal-regulated kinase (pERK) assay in apelin receptor-transfected human embryonic kidney cells. These labeled peptides were then used for both fluorescence and diffusion-ordered nuclear magnetic resonance spectroscopy experiments in the presence of varying concentrations of micelle and bicelle species to test for membrane interaction as required by the membrane catalysis model. Fo¨rster resonance energy transfer experiments were performed with fluorescently labeled apelin isoforms to native tryptophan in fragments of the apelin receptor and to eGFP-tagged full-length apelin receptor to both localize the ligand-receptor binding interface and to quantify the peptide-receptor interaction. 1872-Pos Board B9 Functionality of MscL in Droplet Interface Bilayer Mohammad Heiranian, Amir Barati Farimani, Narayana Aluru. University of Illinois at Urbana-Champaign, Urbana, IL, USA. A droplet interface bilayer (DIB) forms when two water droplets in oil are brought into contact in the presence of lipid molecules. DIBs have shown to be promising in providing a lipid bilayer platform for studying ion channels and biological nanopores. In this work, using extensive all-atom molecular dy- 373a namics simulations (up to 1,000,000 atoms), we study the response of a large conductance mechanosensetive protein channel (MscL), embedded in a DIB, to electrochemical stimulations. MscL channel activation has been known to be tension-dependent meaning that they conduct ions when excited mechanically. Here, we find that a MscL channel at its intermediate expanded state can undergo a conformational change to further expand and reach its fully open state when an ion concentration gradient is applied across the DIB. This gradient is applied such that the salt solution in one droplet is dilute and the other is concentrated. Ions move by diffusion from the high concentrated droplet to the low concentrated droplet resulting in a flow of water and ions through the channel and subsequently expanding MscL. The ion concentration gradient across the DIB introduces a gain of function in MscL opening mechanism and can lead to implementing novel nano-scale biologically-inspired materials and devices. 1873-Pos Board B10 Homology Models of the Trimeric CNG Channel C-Leucine Zipper Domains Offer Insight about the Olfactory CNG Channel Subunit Stoichiometry Dillion M. Fox1, Christopher M. MacDermaid1, Jacqueline Tanaka2. 1 Institute of Computational and Molecular Science, Temple University, Philadelphia, PA, USA, 2Biology, Temple University, Philadelphia, PA, USA. CNG channels in rod, cone and olfactory sensory neurons are tetrameric proteins composed of A-type and B-type subunits. The subunit compositions of rod and cone photoreceptors are 3:1 CNGA1:CNGB1 and CNGA3:CNGB3, respectively. A parallel 3-helix coiled-coil domain in the carboxy-terminal leucine zipper (CLZ) region of CNGA1 constrains the channel to incorporate a single CNGB1 subunit. High-resolution crystal structures of soluble trimeric CLZ domains from CNGA1 and CNGA3 were similar and support the idea that the trimeric CLZ domain governs rod and cone subunit stoichiometry (Shuart et al. 2011 doi:10.1038/ncomms1466). By contrast, olfactory neurons have a subunit composition of 2:1:1 of CNGA2:CNGA4:CNGB1b. We used the X-ray structures of the CNGA1 and CNGA3 CLZ domains to construct a homotrimeric CNGA2 and a hetero-trimeric CNGA2:CNGA4 (2:1) model using probablistic protein design. Once the homology models were constructed, the energies of the structures were minimized to equilibrate the systems. The potential energies of the systems after equilibration serve as a baseline reference point to quantify the overall stability of each structure. The potential energies from CNGA1, CNGA2, CNGA3 and CNGA2/CNGA4 were compared, and the heterotrimer was shown to have a more favorable structure than the homotrimers. We then performed energetic calculations using robust classical methods. The results reinforced the hypothesis that the heterotrimeric CNGA2/CNGA4 CLZ structure is the thermodynamically favored configuration. This approach was used to examine achromatopsia-associated mutations in the CLZ region of the cone CNGA3 channel subunit. The results and implications from the modeling will be presented in the context of achromatopsia pathophysiology. 1874-Pos Board B11 Molecular Dynamics Simulations of Wild-Type and Mutant AQP6 Channels: Investigation of Anion Transport in Human AQP6 Ravi Kumar Verma, Ramasubbu Sankararamakrishnan. Biological Sciences and Bioengineering, Indian Institute of Technology, Kanpur, Kanpur, India. Channels belonging to Major Intrinsic Protein (MIP) superfamily are known to selectively transport water, glycerol and other neutral solutes and inhibit proton and ion transport. Among the 13 known human MIP homologs, AQP6 is the only channel implicated in anion transport. The selectivity filter of AQP6 is identical to AQP1 which is a water selective channel; however, AQP6 shows very little water transport. Additionally, Hg2þ was found to increase water and ion transport through AQP6 channels, which seems counterintuitive as Hg2þ ions inhibits water transport in case of AQP1 by modifying the SF cysteine residue. Previous studies [1] showed that a single mutation, N60G, entirely changes the transport properties of AQP6. N60G mutant was shown to transport water and inhibit anion transport. The current study investigates the effects of N60G mutation on AQP6 water and ion transport using all atom molecular dynamics (MD) simulations and steered molecular dynamics (SMD) approaches. To find out the effect of N60G mutation, we have carried out MD simulations of modelled wild-type and N60G mutant of hAQP6 tetramer in explicit lipid bilayer for 100ns each. MD simulations of the WT and mutant AQP6 channels provide insights into the structural changes that lead to higher water transport in mutant-AQP6. During the simulations no anion was able to traverse the channel completely; however few anions entered the central channel or the monomeric channels indicating the possible paths anions can take. Additionally, comparison of Potential of Mean Force (PMF) profiles of wild-type and mutant-AQP6 374a Tuesday, February 10, 2014 proteins, obtained from SMD simulations, hints towards the unknown mechanism of anion transport through these proteins. [1] Liu et al., PNAS USA 102, 2192-2197 (2005) 1875-Pos Board B12 An Intra-Molecular Disulfide Cross-Link Stabilizes an InwardOriented Transport Intermediate Conformation of the Tonb-Dependent Transporters Shimei Gong1, Nazir Barekzi2, Katarzyna Niedzielska1, Nicholas E. Sherman3, Robert K. Nakamoto1. 1 Molecular Physiology and Biological Physics, University of Virginia, Charlottesville, VA, USA, 2Biological Sciences, Old Dominion University, Norfolk, VA, USA, 3Microbiology, Immunology and Cancer Biology, University of Virginia, Charlottesville, VA, USA. The Gram negative bacteria TonB-dependent transporter (TBDT) BtuB translocates vitamin B12 (cobalamin or Cbl) across the outer membrane. The aminoterminal lumenal domain fits within the 22-stranded ß-barrel and contains several determinants for Cbl binding. Current models suggest that the TBDT carries out active transport driven by energy from the proton motive force (pmf), which is transmitted via direct interactions between the inner membrane protein TonB and the BtuB amino-terminal Ton Box motif. The lumenal domain must undergo large conformational changes to accommodate passage of the substrate through the barrel, but the molecular features of the mechanism are unknown. We show that disulfide bonds can be induced to form in whole cells between three pairs of cysteines introduced at barrel-lumenal domain contact points, consistent with the x-ray crystallographic structures. However, we also find spontaneously formed, stoichiometric cross-links between cysteines in the Ton Box motif and in place of Ser120 that indicate a conformation different from the x-ray structures. The TBDT family members FecA and FhuA with cysteines in equivalent positions also form stoichiometric disulfide bonds. Cbl uptake through BtuB is blocked by the Ton Box Cys-S120C cross-link and activity is recovered upon reduction. Significantly, Cbl binds to the cross-linked BtuB in isolated outer membrane fragments but not to whole cells, indicating that the Cbl binding site of the cross-linked form is oriented towards the periplasmic side. In contrast, the transporter is oriented outwards in all other conditions, included in the absence of a pmf. These results suggest an alternating access transport mechanism. 1876-Pos Board B13 Live-Cell Measurements of the Conformational Rearrangements in Bax at the Initiation of Apoptosis Robert F. Gahl, Yi He, Shiqin Yu, Nico Tjandra. NHLBI-Biochemistry and Biophysics Center, National Institutes of Health, Bethesda, MD, USA. The Bcl-2 family of proteins regulates the activation of apoptosis through the mitochondria pathway. Pro- and anti-apoptotic members of this family keep each other in check until the correct time to commit to apoptosis. The point of no return for this commitment is the permeabilization of the outermitochondrial membrane (OMM). Translocation of the pro apoptotic member, Bax, from the cytosol to the mitochondria is the molecular signature of this event. We employed a novel method to reliably detect Fo¨rster Resonance Energy Transfer (FRET) between pairs of fluorophores to identify intra-molecular conformational changes and inter-molecular contacts in Bax as this translocation occurs in live cells. In the cytosol, our FRET measurements indicated that the C-terminal helix is exposed instead of tucked away in the core of the protein. This coincided with measurements using fluorescence correlation spectroscopy (FCS) that showed that cytosolic Bax diffuses much slower than expected, suggesting possible complex formation or transient membrane interaction. We propose that this exposed helix allows for this contact to occur. Cross-linking the C-terminal helix (a9) to helix a4 reduced the instances of these interactions while at the same time yielded FRET measurements that are consistent with the a9 helix tucked into the core of the protein. After translocation, our FRET measurements showed that Bax molecules form homooligomers in the mitochondria through two distinct interfaces involving the BH3 domain (helix a2) and the C-terminal helix. These findings have implications for possible contacts with other Bcl-2 proteins to create pores to permeabilize the OMM, which would also be necessary for the regulation of apoptosis. 1877-Pos Board B14 FIS1 and DNM1L Cooperate in Mitochondrial Fission: Convergence of Evolution and Intelligent Design Blake Hill1, Megan Cleland Harwig1, Cara Marie Manlandro2, Lora K. Picton3, Nolan W. Kennedy1. 1 Biochemistry, Medical College of Wisconsin, Milwaukee, WI, USA, 2 Chemistry, Johns Hopkins University, Baltimore, MD, USA, 3Biology, Johns Hopkins University, Baltimore, MD, USA. Mitochondrial fission helps to maintain proper mitochondrial homeostasis in a poorly understood manner despite its association with human disease. Several proteins have been identified in this process, but only two – FIS1 and DNM1L – are found in every species that contain mitochondria. We asked whether these proteins might cooperate together in this process. We found that FIS1 has the ability to directly recruit DNM1L, but is auto-inhibited by an N-terminal arm. A point mutant was rationally designed to relieve auto-inhibition and found to increase DNM1L binding and impair mitochondrial fission. This point mutant also increased the population of a latent dimeric state of FIS1 highlighting the complex nature of protein-protein interactions in fission. To address this, we devised an unbiased and general method to rapidly identify residues critical to protein interfaces and applied this technology to yeast Fis1 interactions. Of the >3000 Fis1 alleles screened, ~9% selectively disrupted interactions with one of the three protein partners including DNM1L. To test the functional consequences, each allele was parsed into its corresponding point mutation and tested for mitochondrial fission. Of 211 yeast Fis1 mutants tested to date, 97 resulted in nonfunctional fission indicating that our method identifies residues essential for mitochondrial fission. Orthologous mutations were introduced into human Fis1 and also found to impair interactions with DNM1L and mitochondrial morphology. Analysis of these data supports a new model for the assembly of the mitochondrial fission machinery. 1878-Pos Board B15 Structural Basis for Enhanced Hiv-1 Neutralization by a Dimeric Immunoglobulin G Form of the Glycan-Recognizing Antibody 2G12 Yunji Wu, Pamela J. Bjorkman. California Institute of Technology, Pasadena, CA, USA. The human immunoglobulin G (IgG) 2G12 recognizes high-mannose carbohydrates on the HIV type 1 (HIV-1) envelope glycoprotein gp120. Its two antigenbinding fragments (Fabs) are intramolecularly domain exchanged, resulting in a rigid (Fab)2 unit including a third antigen-binding interface not found in antibodies with flexible Fab arms. We determined crystal structures of dimeric 2G12 IgG created by intermolecular domain exchange, which exhibits increased breadth and >50-fold increased neutralization potency compared with monomeric 2G12. The four Fab and two fragment crystalline (Fc) regions of dimeric 2G12 were localized at low resolution in two independent structures, revealing IgG dimers with two (Fab)2 arms analogous to the Fabs of conventional monomeric IgGs. Structures revealed three conformationally distinct dimers, demonstrating flexibility of the (Fab)2-Fc connections that was confirmed by electron microscopy, small-angle X-ray scattering, and SPR binding studies. We conclude that intermolecular domain exchange, flexibility, and bivalent binding to allow avidity effects are responsible for the increased potency and breadth of dimeric 2G12. In addition, we present this as the first known crystal structure of an IgG dimer. 1879-Pos Board B16 A Computational and Experimental Study of the Structure of FOXl1 Protein Jessica E. Besaw1, Valerie Booth2, Christopher N. Rowley1. 1 Chemistry, Memorial University of Newfoundland, St. John’s, NL, Canada, 2 Biochemistry and Physics & Physical Oceanography, Memorial University of Newfoundland, St. John’s, NL, Canada. Mutated or unregulated FOX proteins have been linked with numerous human genetic diseases. Premature ovarian failure, mental retardation, and severe immune defects are just a few of the severe health problems linked with mutations in the FOXO3a, FOXP1, and FOXN1 proteins, respectively. Studying the structure of FOX proteins is crucial in uncovering the essential structural features that allow these proteins to function properly. However, the structure of the C-terminal domain is unknown for many FOX proteins including FOXL1. In this research, the structure of the C-terminal domain of FOXL1 protein is investigated using both computational and experimental methods. Computationally, first-principle molecular dynamic (MD) folding simulations were performed using replica exchange MD to provide a prediction of the native structure. Experimentally, the C-terminal domain of FOXL1 was expressed, purified, and then structurally characterized using circular dichroism. 1880-Pos Board B17 All-Alpha to All-Beta Structural Conversion in the Transcription Factor RfaH Jeevan B. Gc. Physics, Florida International University, Miami, FL, USA. We used combination of replica exchange molecular dynamics simulations with implicit solvent and detailed all-atom simulations with explicit solvent to investigate the a-helix to b-structure transformation of RfaH-CTD. While interacting with the N-terminal domain (NTD), the C-terminal domain (CTD) of RfaH folds to an a-helix bundle but it undergoes an all-a to all-b Tuesday, February 10, 2014 conformational transformation when it does not interact with the NTD. The RfaH-CTD in the all-a topology is involved in regulating transcription whereas in the all-b topology it is involved in stimulating translation by recruiting a ribosome to an mRNA. Calculations of free-energy landscape and transfer entropy elucidate the details of the RfaH-CTD transformation process. The importance of interfacial interactions between the two domains of RfaH is highlighted by the compromised structural integrity of the helical form of the CTD in the absence NTD. 1881-Pos Board B18 Small-Angle X-Ray Scattering and Biochemical Studies of an Intramolecular Tandem Coiled Coil Donghyuk Shin1, Seungsu Han1, Gwanho Kim1, Gyu Hee Kim1, Xu Xheng2, Yang-Gyun Kim2, Sangho Lee1. 1 Biological Sciences, Sungkyunkwan University, Suwon, Korea, Republic of, 2Chemistry, Sungkyunkwan University, Suwon, Korea, Republic of. Coiled coil has served as an excellent model system for studying protein folding and developing protein-based biomaterials. Most designed coiled coils function as oligomers, namely intermolecular coiled coils. However, less is known about structural and biochemical behavior of intramolecular coiled coils where coiled coil motifs are covalently linked in one polypeptide. Here we prepare a protein which harbors three coiled coil motives with short linkers, termed tandem coiled coil (TCC) and characterize its structural and biochemical behavior in solution. TCC consists of three coiled coil motives whose sequences are derived from Coil-Ser and its domain swapped dimer (DSD). Modifications include positioning E (Glu) residue at ‘‘e’’ and K (Lys) at ‘‘g’’ throughout heptad repeats to enhance ionic interaction among its constituent coiled coil motives. The linkers are four-residue-long with sequence G[S/T] GG to ensure flexibility. Molecular modeling of TCC suggested a compact triple helical bundle structure with the second and the third coiled coil motives forming a canonical coiled coil. TCC exists as a mixture of monomeric and dimeric species in solution. Small-angle X-ray scattering (SAXS) revealed ellipsoidal molecular envelopes for both dimeric and monomeric TCC in solution. The theoretically modeled structures of TCC docked well into the envelopes of both species. Higher ionic strength shifted the equilibrium into monomer with apparently more compact structure. Secondary structure of TCC at various ionic strengths remains unchanged probed by circular dichroism. Taken together, our results suggest that our designed TCC is predominantly monomeric structure through the enhanced ionic interactions, and it is affected by the concentration of ionic species in the buffer. 1882-Pos Board B19 Characterization of Amynthas Gracilis Hemoglobin (HbAg) and its Subunits by AUC and MALDI-TOF-MS Patricia S. Santiago1, Francisco Adriano O. Carvalho2, Jonathan B.S. Oliveira1, Angela P.D. Linhares1, Patrı´cia G. Morgante1, Jose´ Wilson P. Carvalho2,3, Marcel Tabak2. 1 Agronomy, UNESP/Registro, Registro, SP, Brazil, 2Molecular Physical Chemistry, Instituto de Quı´mica de Sa˜o Carlos, USP, Sa˜o Carlos, SP, Brazil, 3 Universidade do estado do Mato Grosso, UNEMAT, Barra do Bugres, Brazil. The giant extracellular hemoglobin (HbAg) of the annelid Amynthas gracilis has a molecular mass (MM) of 3400kDa. In the current work, the characterization of MM values of HbAg and its subunits is presented. Electrophoresis, MALDI-TOF-MS and AUC show that the MM values of HbAg subunits are very close, but not identical to those of Glossoscolex paulistus (HbGp) and Rhinodrilus alatus (HbRa) hemoglobins. Analytical ultracentrifugation (AUC) sedimentation velocity experiments were performed to obtain M for HbAg in oxy- form. value of 59.3 5 0.2 S was obtained for native HbAg. From the ratio between sedimentation and diffusion coefficients values for M of approximately 3400 5 100 kDa for oxy-HbAg was obtained. MALDITOF-MS data gave MM for HbAg subunits. Monomer d is found to exist in, at least, four isoforms with MM 16,24453Da, 16,45955Da, 16,66755Da and 16,85553Da, as noticed for HbGp, and not observed for HbRa. Furthermore, the trimer subunit presents two isoforms ((abc)1 and (abc)2) with MM 51,415520Da and 51,610514Da, respectively. This might indicate that the monomers a, b and c do have isoforms, as found for HbGp and not for HbRa. The monomeric chains a, obtained from the trimer abc reduction, present three isoforms with MM 17,015Da, 17,061Da and 17,138Da, differing from HbGp that presents four isoforms. A less intense species is observed at 67,717, and is due to the tetramer abcd contribution. Finally, AUC and MALDI-TOF-MS data are very close as compared to that obtained for HbGp and HbRa. Our results show total consistency between M obtained by AUC and recent partial characterization by mass spectrometry. Finantial support: FAPESP and CNPq Brazilian agencies. 375a 1883-Pos Board B20 Structure and Function of Clostridial Yter Margaret Hurley1, Katherine L. Germane2, Matthew Servinsky3, Elliot Gerlach4, Christian Sund3. 1 US Army Research Laboratory, Aberdeen Proving Ground, MD, USA, 2Oak Ridge Associated Universities, Belcamp, MD, USA, 3US Army Research Laboratory, Adelphi, MD, USA, 4Federal Staffing Resources, Annapolis, MD, USA. Pectin found in fruit and vegetable waste is a potential renewable feedstock for production of butanol from Clostridium acetobutylicum. Pectin is an abundant complex carbohydrate found in the cell wall of plants, but its fermentation results in low butanol yields. Understanding the process of degradation and metabolism of the carbohydrate matrix is key to improving butanol yields from pectin. Transcriptomic analysis of C. acetobutylicum during growth on pectin identified several genes with potential degradation activity. One of these genes, CA_C0359, encodes a putative unsaturated rhamnogalacturonyl hydrolase (URH). The crystal structure of the recombinant CA_C0359 protein was ˚ resolution. We present here an overview of the crystal strucsolved to a 1.6 A ture of the CA_C0359 protein, and theoretical results docking various disaccharides into this structure to assess its capabilities for sugar degradation in the context of functionally similar proteins. 1884-Pos Board B21 NMR Structural Characterization for Proteases of Dengue and West Nile Viruses and its Insight into Drug Discovery Congbao Kang. Experimental Therapeutics Centre, Agency for Science, Technology and Research, Singapore, Singapore. Flaviviruses are a major cause of infectious disease in human, which include the Dengue Virus (DENV), West Nile virus (WNV). The genomic RNA encodes a polyprotein precursor which is processed proteolytically upon translation to 10 proteins, including three structural proteins (capsid [C], premembrane [prM] and envelope [Env]), and seven nonstructural (NS) proteins (NS1, NS2A/B, NS3, NS4A/B, and NS5). The NS3 is of great interest in drug discovery because of its N-terminal domain has the protease activity required for viral replication. The activity of NS3 is regulated by membrane protein NS2B. Previous studies were using a construct that contains 40 amino acids from NS2B fused with NS3 protease domain though an artificial G4SG4 linker. We have developed a series of peptidic inhibitors targeting WNV and DENV proteases. We analyzed their interactions with the protease using both chemical shift perturbation and docking studies. For the DENV protease, there is still no potent inhibitor available so far. Using NMR spectroscopy, we discovered that the conventional DENV protease construct in which NS2B cofactor region linked with NS3 protease through a flexible liner is not suitable for drug discovery due to the protein dynamics. Using a co-expression system, we obtained a protease complex that contains the NS3 protease domain and 50-residue segment of the NS2B. Our results show that this protease complex exists as a close conformation and active under physiological conditions, which might be a better construct for drug discovery targeting DENV. We also expressed and purified a natural form of DENV protease which was shown to be active in detergent micelles. Our studies will be useful for development of DENV protease inhibitors. 1885-Pos Board B22 Monitoring Protein Structure on the Surface of Gold Nanoparticles using NMR Spectroscopy Ailin Wang, Karen Woods, Tam Vo, Alex Coats, Nicholas C. Fitzkee. Department of Chemistry, Mississippi State University, Mississippi State, MS, USA. Because of their unique spectroscopic properties and biocompatibility, proteinfunctionalized gold nanoparticles (AuNPs) have many potential diagnostic and therapeutic applications. Surface-bound proteins can be used both as molecular sensors and drug delivery vectors, and the plasmonic properties of gold enable the detection of very small changes in surface chemistry. Unfortunately, the design of general-purpose, functionalized AuNPs is severely complicated by our limited understanding of protein structure on nanoparticle surfaces. Some enzymes remain active on AuNPs while others are inactive, and it is currently impossible to predict which behavior will be observed. To address this problem, we have recently developed several new NMR-based approaches for monitoring protein structure on AuNP surfaces. We find that the adsorption capacity of 15 nm AuNPs can be predicted using the native structure, suggesting that proteins remain globular on the AuNP surface. Additionally, we demonstrate that proteins can be displaced from AuNPs by organothiols, supporting a case for reversible binding. Finally, we have used hydrogen/deuterium exchange (HDX) to monitor structural perturbations of two model proteins, 376a Tuesday, February 10, 2014 GB3 and Ubiquitin, when bound to AuNPs. We find no significant changes in slow HDX rates (5-300 min), suggesting that AuNP-induced structural changes are small for these two proteins. Together, these results support a model where most of a protein’s native contacts are preserved upon adsorption, although larger changes may occur over long timescales. 1886-Pos Board B23 3D Reconstruction of the S885A Mutant of the Human Mitochondrial Lon Protease Sami Kereiche, Lubomir Kovacik. Institute of Cellular Biology and Pathology, 1st faculty of Medicine, Charles University in Prague, Prague, Czech Republic. The Lon protein is a protease belonging to the superfamily of ATPases Associated with diverse cellular Activities (AAAþ). Its main function is the control of protein quality and the maintenance of proteostasis by degrading misfolded and damaged proteins, which occur in response to numerous stress conditions. Lon protease has been also shown to participate in regulation of levels of transcription factors that control pathogenesis, development and stress response. Furthermore, it seems to play an important role in aging, and it is supposed to be involved in mtDNA replication, translation, or repair. We focus our interest on the structure of human mitochondrial Lon (hLon) protease whose altered expression levels are linked to some severe diseases, such as epilepsy, myopathy, or lateral sclerosis. At the moment, it is assumed that Lon subunits assemble into oligomeric structures whose conformations are supposed to differ at ATP, ADP, and protein substrate binding. However, neither the full 3D structure of the Lon holoenzyme nor the mechanism of Lon’s action is known. Several sub-structures of bacterial and human Lon have been resolved by X-ray scattering, and one 3D structure of an E. Coli Lon dodecamer active at physiological protein concentrations was resolved with electron microscopy. Here, we present two conformations of an ADP-bound hLon S885A mutant obtained as a result of cryo-EM data analysis. The S885A mutant has a point mutation on the proteolytic domain, which completely disables its proteolytic function but does not affect its ATP-binding properties. The 3D reconstructions reveal that human Lon is a hexamer whose proteolytic and ATPase domains are arranged into a helix. The opening and pitch of the helix depend on the N-terminal domain interactions. These structures provide an insight toward the understanding of the protein mechanism of action. 1887-Pos Board B24 Structure and Dynamics of the EIIC Sugar Uptake System Zhenning Ren, Ming Zhou. Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, USA. The phosphoenolpyruvate-dependent carbohydrate phosphotransferase system (PTS) is a sugar uptake system unique to bacteria. It is a multicomponent system consisting of several cytosolic proteins and a dimeric transmembrane protein (EIIC in most PTS systems), which transports extracellular sugar across the membrane. Although EIIC is a uniporter, it is able to drive concentrative transport of its ligand because the sugar is phosphorylated by a cytosolic protein, EIIB, while still bound to the transporter. Phosphorylation prevents the sugar from escaping the cell and primes it for consumption by the cell. Little is known regarding the mechanism of sugar translocation and phosphorylation. Currently, the only available crystal structure of an EIIC is that of the N,N0 -diacetylchitobiose transporter, bcChbC (1). We have proposed a mechanism for sugar translocation and phosphorylation, and we will report our progress in characterizing the mechanism. Reference 1. Cao Y, Jin X, Levin EJ, et al. Crystal structure of a phosphorylation-coupled saccharide transporter. Nature. 2011;473(7345):50-4. 1888-Pos Board B25 Predicting the Effects of Clinically Observed Kinase Mutations using Molecular Modeling and Machine Learning Algorithms E. Joseph Jordan1, Peter J. Huwe1, Yael Mosse2, Mark Lemmon1, Ravi Radhakrishnan1. 1 The University of Pennsylvania, Philadelphia, PA, USA, 2Children’s Hospital of Pennsylvania, Philadelphia, PA, USA. Many cellular processes are impacted by signaling through receptor and nonreceptor kinase proteins. These include such diverse cellular actions as proliferation, differentiation, and motility, as well as tissue level phenomena such as angiogenesis and development. This important role in the cell is reflected also in the relative overrepresentation of kinases among known cancer mutations to proteins. In order to better understand the functional effects of these mutations, we have developed computational methods that seek to predict the effect of point mutations on kinase activation. By predicting whether a given mutation causes a kinase to be more active, we can gain insight into the overall impact of the mutation on cell phenotype and give insight to clinicians on patient cohorting for efficacious treatment with targeted kinase inhibitors. We have developed two separate but complementary methods to predict kinase activation status. The first uses molecular dynamics (MD) simulations and scoring criteria to predict if a mutation preferentially stabilizes the protein’s active state. As a complimentary approach to MD, we have developed machine learning techniques that utilize the method known as support vector machines to predict whether mutations in a large number of kinases (>450) are activating. This method has proven to be almost as effective at predicting activation mutations as the mechanistic picture gained from MD simulations. We think these methods are both broadly applicable and have the potential to greatly impact both our understanding of mechanisms of kinase activation as well as to guide best practices in the clinical setting of targeted therapy in cancer treatment. 1889-Pos Board B26 Activation Mechanism of a Signaling Protein at Atomic Resolution Francesco Pontiggia1, Dimitar V. Pachov1, Michael W. Clarkson1, Janice Villali1, Michael F. Hagan2, Vijay S. Pande3,4, Dorothee Kern1,5. 1 Department of Biochemistry, Brandeis University, Waltham, MA, USA, 2 Department of Physics, Brandeis University, Waltham, MA, USA, 3 Department of Chemistry, Stanford University, Stanford, CA, USA, 4 SIMBIOS, NIH Center for Biomedical Computation, Department of Bioengineering, Stanford University, Stanford, CA, USA, 5Howard Hughes Medical Institute, Waltham, MA, USA. The interconversion between the inactive and active state is the heart of signaling. This process has traditionally been described by the two corresponding structures, sometimes complemented with kinetic data. However the question of how these folded states interconvert is largely unknown due to the inability to experimentally observe the transition pathways. Here we present a recent investigation of the full free energy landscape of the receiver domain of the response regulator NtrC (NtrC ) by combining several computational methods including the string method, Markov state models of massive unbiased MD simulations, and long MD simulations on ANTON, with new NMR structural data. The results unveil several unexpected features underlying efficient signaling: The active and inactive states have to be considered purely in kinetic terms. The functional need of attaining a stable and well-defined conformer, crucial to the active form of the protein, is absent in the inactive state. The inactive state comprises a structurally heterogeneous collection of sub-states that interconvert on timescales shorter than the transition to the active state. The transitions between the two functional states occur through multiple pathways characterized by transition states with dramatically different structural features. In addition to this entropic lowering of the transition barrier, a number of polar side-chains engage in unspecific transient interactions during the barrier crossing and thus make the activation mechanism flexible, efficient and robust. These novel findings challenge the structural paradigm of signaling and may represent general features for functional conformational transitions within the folded state. 1890-Pos Board B27 Crystal Structures of Trehalose Synthase from Deinococcus Radiodurans Reveal a Closed Conformation for Intramolecular Isomerization Catalysis and Mutant Induction of an Active-Site Aperture Sih-Yao Chow1, Yung-Lin Wang2, Li-Ci Ye1, Shwu-Huey Liaw1. 1 Institute of Genome Sciences, National Yang Ming University, Taipei City, Taiwan, 2Institute of Biochemistry and Molecular Biology, National Yang Ming University, Taipei City, Taiwan. Trehalose has been used in food, cosmetic, and biotechnological industries due to its exceptional stability. Trehalose synthase (TS) catalyzes a simple conversion of inexpensive maltose into trehalose and hence has a great potential. TS consists of a catalytic (b/a)8 barrel, a subdomain B, a C-terminal b domain and two TS-unique subdomains (S7 and S8). The apo TS structures from Mycobacterium smegmatis and M. tuberculosis showed an unusual inactive conformation, in which the S7 loop blocks the substrate-binding pocket. Here we report structural and mutational studies of TS from Deinococcus radioduran (DrTS). The complex structures of DrTS with the inhibitor Tris share high homology with the substrate-bound sucrose hydrolase, amylosucrase, and sucrose isomerase, particularly virtually identical active-site architectures. A maltose was modelled into the active site and subsequent mutational analysis suggested that Tyr213, Glu320, and Glu324 are essential for the TS activity. In addition, the interaction networks between subdomains B and S7 seal the active-site entrance. Disruption of such networks through replacement of Arg148 and Asn253 with alanine resulted in a decreased isomerase activity but an increased hydrolase activity. The R148A and N253A structures showed a small pore created for water entry. Tuesday, February 10, 2014 Unexpectedly, the apo N253F mutant with ~80% of hydrolase activity but no detectable isomerase activity showed a strikingly different conformation, in which the Bb1-Bb2 loop in subdomain B is disordered, and the subdomain B rotates away to create an open active site. Interestingly, this mutant displays a high structural similarity to the apo sucrose hydrolase. Therefore, our DrTS-N253F structure may represent an open conformation for the apo TS, while the DrTSTris may represent a substrate-induced closed conformation that will facilitate intramolecular isomerization and minimize disaccharide hydrolysis. 1891-Pos Board B28 Biophysical Characterization of Naturally Occurring Titin-M10 Mutations Nathan T. Wright, Michael W. Rudloff. Chemistry and Biochemistry, James Madison University, Harrisonburg, VA, USA. The extreme C-terminus of titin (termed the M10 domain) binds to the N-terminus of obscurin in the M-band of skeletal muscle cells. Multiple M10 mutations are linked to limb-girdle muscular dystrophy type 2J (LGMD2J) and tibial muscular dystrophy (TMD) in humans. The high-resolution structure of M10 has been solved, along with M10 bound to an obscurin-like target. However the effect of the M10 mutations on protein structure and binding has not been thoroughly characterized. Here we express all four of the naturally occurring human M10 missense mutants and biophysically catalogue them. Three of the four mutations are severely misfolded, and are binding incompetent. One mutation, I57N (also called the Belgian mutation), shows no significant structural, dynamic, or binding differences from the wild-type domain. We suggest that this mutation is not directly responsible for muscle wasting disease, but is instead merely a silent mutation found in symptomatic patients. Protein Dynamics and Allostery II 1892-Pos Board B29 Investigating the Mechanism of Iron Dependent Repressor (IDER) Activation and DNA Binding Soma Ghosh1, Nagasuma Chandra2, Saraswathi Vishveshwara3. 1 I.I.Sc. Mathematics Initiative, Indian Institute of Science, Bangalore, India, 2 Department of Biochemistry, Indian Institute of Science, BANGALORE, India, 3Molecular Biophysics Unit, Indian Institute of Science, Bangalore, India. Metalloproteins form a major class of enzymes in the living system and are involved in critical biological functions such as catalysis, redox reactions and as ‘‘switches’’ in signal transductions. Iron dependent repressor (IdeR) is a metal-sensing transcription factor that regulates free iron concentration in Mycobacterium tuberculosis. IdeR is also known to promote bacterial virulence, making it an important protein for therapeutics. In this study, we have employed molecular dynamic simulations on different binding states of IdeR in the presence and absence of iron to study its influence on protein function. Structures were investigated using hydrogen bonds and protein structure networks and displayed significant variation between the metallated and the non-metallated systems. Briefly, we could establish the role of iron in stabilizing the monomeric unit of IdeR which in turn promotes protein dimerization. Two major monomer conformations, ‘‘open’’ and ‘‘closed’’ were identified and their geometrical parameters were also quantified. Perhaps, the most striking results are obtained from the simulations of the IdeRDNA complex in the absence of metals, where the protein subunits are seen to dissociate away from the DNA quite rapidly. Such drastic changes in the IdeRDNA interactions not only provide molecular insights about the role of iron, but also about the mechanism of DNA binding and unbinding. Based on the ensemble structure analysis, we suggest the role of iron as a possible allosteric effector that enhances the IdeR-DNA interactions. Our simulation results enable us to understand the sequence of events that govern IdeR-DNA binding in the presence of iron. 1893-Pos Board B30 Dynamic Characteristics of Allosteric Pathways in scFv Antibody Fragments Amit Srivastava1, Malgorzata B. Tracka2, Shahid Uddin2, Jose Casas-Finet3, Dennis R. Livesay1, Donald J. Jacobs4. 1 Department of Bioinformatics and Genomics, University of North Carolina, Charlotte, NC, USA, 2Formulation Sciences, MedImmune Ltd., Cambridge, United Kingdom, 3Analytical Biochemistry, MedImmune LLC, Gaithersburg, MD, USA, 4Dept. of Physics and Optical Science, University of North Carolina, Charlotte, Charlotte, NC, USA. Proteins exhibit dynamic behavior that is constrained by the hydrogen bond network (HBN). Previous work [Tong Li, et. al. PLoS ONE 9(3) 2014] on a set of six single chain Fv (scFv) anti-lymphotoxin-b receptors demonstrated that there is a redistribution of flexibility upon mutation due to changes in the HBN. The observed redistribution occurs due to enthalpy-entropy compen- 377a sation in the native state ensemble. Moreover, the shifts in rigidity and flexibility follow the Le Chaˆtelier’s principle, meaning increased rigidity is offset by increased flexibility elsewhere. Extending this work further, the thermodynamic and mechanical response is calculated for localized mechanical perturbations that reduce conformational entropy along the protein backbone. At each mechanical perturbation site all other residues that have significant changes in flexibility are identified. Some perturbation sites yield no statistically significant response, and others yield a response that is spatially localized near the perturbation site. A relatively small fraction of perturbations generate strong distal responses, indicating they are putative allosteric sites. Importantly, the allosteric pathways that carry the distal changes in flexibility or rigidity are linked to fluctuations in the HBN, which also depend on the redistributions of rigidity and flexibility that occur upon mutation. Mutations induce a population shift that changes the most probable constraint networks in the equilibrium ensemble, and alter the mechanical signaling pathway through the modification of the HBN. Interestingly, a reciprocal relation is observed among conjugate response and perturbation sites, such that they can be interchanged in their role. A comparative analysis on response maps due to perturbation is made across all six mutant structures, which provide important insight into how sensitive allosteric mechanisms are within antibody fragments. 1894-Pos Board B31 Functionally Important Residues from Mode Coupling during Short-Time Protein Dynamics Onur Varol1, Deniz Yuret2, Burak Erman3, Alkan Kabakcioglu4. 1 Informatics, Indiana University, Bloomington, IN, USA, 2Computer Science, Koc University, Istanbul, Turkey, 3Chemical and Biological Engineering, Koc University, Istanbul, Turkey, 4Physics, Koc University, Istanbul, Turkey. Relevance of mode coupling to energy/information transfer during protein function, particularly in the context of allosteric interactions is widely accepted. However, existing evidence in favor of this hypothesis comes essentially from model systems. We here report a novel formal analysis of the near-native protein dynamics which allows us to explore the impact of the interaction between (possibly non-Gaussian) vibrational modes on fluctutational dynamics. We show that, an information-theoretic measure based on mode coupling alone yields a ranking of residues with a statistically significant bias favoring the functionally critical locations identified by experiments. 1895-Pos Board B32 High-Speed AFM Observation of Antibody IGG Characteristic of Swinging Arms Norito Kotani, Tomohiro Hirano, Takashi Morii, Takao Okada. Biomolecule Metrology, Research Institute of Biomolecule Metrology, Tsukuba, Japan. Antibody IgG molecule is a ‘‘Y’’ shape protein. It has two Fab regions and one Fc region. Fab regions bind to the antigens. Hinge region connects a Fab region to the Fc region. High-speed AFM (HS-AFM), developed by Prof. Ando in Kanazwa University, can observe dynamic behavior of motor protein, myosin as movie without chemical fixing or stain treatment (1, 2). We observed IgG in solution using HS-AFM. ‘‘Y’’ shape of IgG was imaged clearly, and Fab and Fc regions were distinguished. The Fab regions moved in torsional direction like swinging arms. This behavior depends on flexible structure of hinge regions. We analyzed the Fab swivel movements as random walks, and estimated the flexibility of the IgG hinge region. The flexible nature of hinge region contributes for the antibody to bind to the antigen. For the first time, we have identified the swinging nature of this soft structure, which is important for antibody function. The lacking of swing movement would lead to reduce binding between antibody and antigen (3). HS-AFM can directly observe dynamic behaviors of biomolecules as movie in solution, and reveal functions in detail. 1. T. Ando et al., Proc. Natl. Acad. Sci. USA. 98, 12468- (2001). 2. N. Kodera et al., Nature 468: 72- (2010) 3. J. Preiner et al., Nature Communications 5: 4394- (2014). 1896-Pos Board B33 Visualizing Global Properties of a Molecular Dynamics Trajectory Hao Zhou1, Shangyang Li1, Makowski Lee2,3. 1 Department of Electrical and Computer Engineering, Northeastern University, Boston, MA, USA, 2Department of Bioengineering, Northeastern University, Boston, MA, USA, 3Department of Chemistry and Chemical Biology, Northeastern University, Boston, MA, USA. Molecular dynamics (MD) trajectories are very large data sets that contain substantial information about the dynamic behavior of a protein. Condensing these data into a form that can provide intuitively useful understanding of 378a Tuesday, February 10, 2014 the molecular behavior during the trajectory is a substantial challenge that has received relatively little attention. Here, we introduce the sigma-r plot, a plot of the standard deviation of intermolecular distances as a function of that distance. This representation of global dynamics contains within a single, onedimensional plot, the average range of motion between pairs of atoms within a macromolecule. Comparison of sigma-r plots calculated from 10 nsec trajectories of proteins representing the four major SCOP fold classes indicates significant diversity of dynamic behaviors which are recognizably different among the four classes. Differences in domain structure and molecular weight also produce recognizable features in sigma-r plots, reflective of differences in global dynamics. Plots generated from trajectories with progressively increasing simulation time reflect the increased sampling of the structural ensemble as a function of time. Single amino acid replacements can give rise to changes in global dynamics detectable through comparison of sigma-r plots. Dynamic behavior of substructures can be monitored by careful choice of interatomic vectors included in the calculation. Comparison between the sigma-r plots calculated from MD simulations and from wide angle x-ray solution scattering data is also feasible with the potential for providing direct experimental tests of the approximations required for coarse-grained MD simulations. These examples provide demonstrations of the utility of the sigma-r plot to provide a simple measure of the global dynamics of a macromolecule. 1897-Pos Board B34 Computational Modeling of the FcaRI Receptor Binding in the Fca Domain of the Human Antibody IgA: Corse-Grained Molecular Dynamics (MD) Methods Manori Jayasinghe1, Monica T. Posgai2, Sam Tonddast-Navaei3, George M. Ibrahim4, George Stan3, Andrew B. Herr5. 1 Mathematics physics and computer science, University of Cincinnati Blue ash college, Blue Ash, OH, USA, 2School of Medicine, University of Cincinnati, Cincinnati, OH, USA, 3Department of Chemistry, University of Cincinnati, Cincinnati, OH, USA, 4Department of Medical Sciences, University of Toronto, Toronto, Toronto, ON, Canada, 5Department of Molecular Genetics, Biochemistry & Microbiology, University of Cincinnati college of Medicine, Cincinnati, OH, USA. FcaRI receptor binding in the Fca domain of the antibody IgA triggers immune effector responses such as phagocytosis, antibody-dependent cell-mediated cytotoxicity, respiratory burst and cytokine release in eukaryotic cells. Fca is a dimer of heavy chains of the IgA antibody and each Fca heavy chain which consisted of two immunoglobulin constant domains, CH2 and CH3, can bind one FcaRI molecule at the CH2-CH3 interface forming a 2:1 stoichiometry which is unique to the human IgA. Experimental evidences confirmed that FcaRI binding to the Fca CH2-CH3 junction altered the kinetics of HAA lectin binding at the distant IgA1 hinge and distant Fab region. Given the importance of residues near the CH2-CH3 junction for receptor binding that were predicted experimentally by binding energetic analysis, our focus in this computational research was to understand the conformational changes and the residue-pairs in long-range communication which co-ordinate the receptor binding dynamics of the Fca dimer complex. We computed the principal collective motions by using the corse-grained structure based molecular dynamics trajectories performed on the high resolution crystal structure of Fca-FcaRI 2:1complex of PDB ID 1OW0 to understand the functional dynamics in Fca. We used three distinct Fca conformations namely free Fca, Fca-FcaRI 1:1 asymmetric and Fca-FcaRI 2:1 symmetric complexes to comparatively study the functional dynamics induced upon receptor binding. Our findings confirmed that FcaRI binding, either in asymmetric or symmetric complex with Fca, propagated long-range conformational changes across the Fc domains, potentially also impacting the hinge and Fab regions. Key words: IgA antibody, single-basin structure-based coarse grain MD simulation, principal component modes, long-range interaction, ligand-induced conformational changes 1898-Pos Board B35 Computer-Aided Drug Discovery Approach Finds Calcium Sensitizer of Cardiac Troponin Steffen Lindert1, Monica X. Li2, Brian Sykes2, J. Andrew McCammon1. 1 UCSD, La Jolla, CA, USA, 2University of Alberta, Edmonton, AB, Canada. Defects in the contractile machinery can lead to heart failure. Weakened contraction of the heart will lead to diminished blood supply of the organs in the human body. Thus, in the fight against heart failure, therapeutics that have the ability to increase the contractile power of the heart are urgently needed. One possible route of action to improve heart contractile power is increasing the calcium sensitivity of the thin filament. From a pharmaceutical standpoint, calcium sensitizers have the distinct advantage of not altering cardiomyocyte calcium levels and thus have lower potential for side effects. Small chemical molecules have been shown to bind to the interface between cTnC and the cTnI switch peptide and exhibit calcium sensitizing properties, possibly by stabilizing cTnC in an open conformation. Building on existing structural data of a known calcium sensitizer bound to cardiac troponin, we devised a combined computational and experimental drug discovery approach. We used Molecular Dynamics to sample a range of troponin structure conformations and accounted for receptor flexibility by running virtual screens into several conformational states. The most promising compounds were then tested using solution NMR titration assays. We were able to identify a novel calcium sensitizer 4-(4-(2,5-dimethylphenyl)-1-piperazinyl)-3-pyridinamine (NCI147866) which binds to cTnC and the cTnCcTnI147-163 complex. Its presence increased the affinity of switch peptide to cTnC by approximately a factor of two. This action was comparable to that of known levosimendan analogues and served as an excellent starting point for targeted compound improvement aimed at higher affinity and calcium sensitization. 1899-Pos Board B36 A Coarse-Grained Langevin Equation for Protein Dynamics: Global Anisotropy and a Mode Approach to Local Complexity Jeremy T. Copperman, Marina G. Guenza. Physics, University of Oregon, Eugene, OR, USA. We utilize a multi-scale approach where molecular dynamic simulations are performed to obtain quantitative structural averages used as input to a coarsegrained Langevin Equation for Protein Dynamics, which can be solved analytically. The approach describes proteins as fundamentally semiflexible objects collapsed into the free energy well representing the folded state. The normal mode analytical solution to this Langevin equation naturally separates into global modes describing the fully anisotropic tumbling of the macromolecule as a whole, and internal modes which describe local fluctuations about the folded structure. Complexity in the configurational free energy landscape around the folded state of the macromolecule leads to a renormalization of the internal modes, while the global modes provide a basis set in which the dipolar orientation and global anisotropy can be accounted for when comparing to experiments. Fundamental to this approach is the inclusion of internal dissipation which is absent in any rigid-body hydrodynamical modeling scheme. This simple approach predicts the dynamics of both global rotational diffusion and internal motion from the picosecond to the nanosecond regime, and is quantitative when compared to time correlation functions calculated from molecular dynamic simulations and in good agreement with Nuclear Magnetic Resonance relaxation experiments. Results for several well-characterized globular proteins are presented, suggesting our method describes the relevant dynamics around the global minimum well. Use of non-equilibrium simulation techniques such as metadynamics to sample the full free-energy landscape of the protein, and extension of the theoretical treatment to describe the dynamics into the biologically interesting microsecond to millisecond regime, will be discussed. 1900-Pos Board B37 Looking at Estrogen Receptor from Small Angles Sichun Yang, Wei Huang, Krishna M. Ravikumar. Center for Proteomics and Department of Pharmacology, Case Western Reserve University, Cleveland, OH, USA. The estrogen receptor (ERa) functions as a hormone-activated transcription factor. The protein is multidomain and highly flexible. To date, however, it remains unclear how various domains interact with one another within the functional ER homodimer. Here, we show via a computational-experimental study that binding of ligand and DNA can allosterically act on the ER’s domain-domain organizations and interactions. First, a set of putative conformations are identified from enabling simulations that search exhaustively all possible domain-domain interactions. Second, multiple major conformations are identified on the basis of experimental synchrotron-based measurements using SAXS and footprinting data that are best-interpreted by computational results from simulations. Finally, data from chemical cross-linking are used to verify the identified ER conformations in solution. This tight integration of multi-technique measurements provides unique insight into the function of ER that dynamically changes its conformations in response to ligand and DNA binding, both of which play critical roles in the development and progression of breast cancer. 1901-Pos Board B38 Study of Proton Transfer in Escherichia Coli Photolyase Meng Zhang1, Zheyun Liu2, Jiang Li3, Lijuan Wang3, Dongping Zhong3. 1 Biophysics, The Ohio State University, Columbus, OH, USA, 2Chemistry and Biochemistry, The Ohio State University, Columbus, OH, USA, 3 Physics, The Ohio State University, Columbus, OH, USA. Photolyase is a flavoenzyme which utilizes blue-light energy to repair UVlight damaged DNA. The catalytic cofactor of photolyase, flavin adenine Tuesday, February 10, 2014 dinucleotide (FAD), has five redox states. Conversions between these redox states involve intraprotein electron transfer and proton transfer, which play important role in protein function. We systematically studied proton transfer in E. coli photolyase in vitro by site-directed mutagenesis and steady-state UV-visible spectroscopy, and proposed the proton channel in photolyase for the first time. We found that in the mutant N378C/E363L, proton channel was completely eliminated when DNA substrate was bound to the protein. Proton is suggested to be transported from protein surface to FAD by two pathways: the proton relay pathway through E363 and surface water to N378 and then to FAD; and the proton diffusion pathway through the substrate binding pocket. In addition, reaction kinetics of conversions between the redox states was then solved and redox potentials of the redox states were determined. These results described a complete picture of FAD redox changes in E. coli photolyase, which are fundamental to the functions of all flavoenzymes. 1902-Pos Board B39 Abrogating Ras Abnormal Function by Targeting Membrane Bound Ras Monomers and Oligomers Priyanka Prakash Srivastava, Alemayehu A. Gorfe. Integrative Biology and Pharmacology, University of Texas Health Science Center at Houston, Houston, TX, USA. Ras is a lipid-modified GTPase that acts as a molecular switch by cycling between active and inactive conformational states and is involved in a plethora of cell signaling pathways. Somatic mutations in Ras are associated with a variety of cancers and are found in ~15% of human tumors such as pancreatic, colorectal, lung, breast cancer to name a few. Of the three major human Ras isoforms H-, N- and K-Ras, cancers associated with mutant K-Ras are the most lethal. Characterization and targeting of hot-spot residues required for the dynamic assembly of Ras on the plasma membrane could be of therapeutic relevance and may yield isoform-specific drugs. To this end, we performed microsecond-level atomistic molecular dynamics (MD) simulations of fulllength, oncogenic K-Ras monomers bound to a heterogeneous membrane. We also carried out extensive protein-protein docking combined with allatom MD to determine the homo-dimeric interface of the protein. MDderived populations from the monomer simulations reveal K-Ras residues interacting directly with the membrane, predominantly in two different modes. Different docking approaches resulted in the identification of several dimer models. In silico mutagenesis and MD-optimization of these models in membrane revealed hot-spot residues that likely form the dimer interface. We will discuss these results in terms of their potential usefulness for anti-cancer drug design. 1903-Pos Board B40 Identifying Transient Binding Pockets in Protein Dynamics for Allosteric Drug Design Supriyo Bhattacharya, Vinod Kasam, Hubert Li, Nagarajan Vaidehi. Immunology, City of Hope National Medical Center, Duarte, CA, USA. Allosteric modulators that regulate the activity of the orthosteric ligands are emerging as cutting-edge strategies in drug design. Unlike orthosteric ligands, allosteric modulators bind to topographically distinct domains from those utilized by orthosteric ligands. Allosteric modulators offer unique therapeutic advantages such as high selectivity thereby causing reduced side effects. However, allosteric pockets are difficult to find since they are often formed transiently during the protein dynamics and hence could be absent in the crystal structures. This poses a challenge in designing allosteric modulators using structure based drug design methods that rely solely on crystal structures or homology models. Moreover not all transient pockets are suitable for allosteric modulation, since the allosteric pocket must communicate with the orthosteric site for functional modulation. Thus there is a dire need for novel techniques that utilize information from protein dynamics to detect allosteric sites for drug design. We present here a comprehensive method for designing allosteric modulators using protein dynamics trajectories or NMR data. We have developed a method, VoidVol, to identify transient binding cavities during protein dynamics. Next, using mutual information calculated from the dynamics trajectories, we map the allosteric pipelines communicating with the orthosteric site. The transient pockets having strong allosteric communication with the orthosteric site can be used for screening allosteric modulators. These sites can be further tested for druggability using the program FindBindSite, also developed in our laboratory. The resulting druggable sites can then be used for high-throughput screening of small-molecule database. We have validated this approach using several kinases and GPCRs with known allosteric modulators. The above methodology demonstrates how molecular dynamics can be useful for allosteric drug design. Our method is applicable to any watersoluble or membrane protein with an available crystal structure or homology model. 379a 1904-Pos Board B41 Learning about Transitions: Adaptive Control in the Molecular Marshal (M2) Framework Thomas B. Woolf1, Sarana Y. Nutanong2, Yanif Ahmad3, Raman Arora3. 1 Physiology, Biophysics and Biophysical Chemistry and Computer Science, Johns Hopkins University, Baltimore, MD, USA, 2Computer Science, City University of Hong Kong, Hong Kong, Hong Kong, 3Computer Science, Johns Hopkins University, Baltimore, MD, USA. Improvements in sampling on the events leading to transitions can provide significant insights into what drives biomolecular change. We present additions to our Molecular Marshal (M2) software framework for the adaptive sampling of biomolecular transitions. As an example, we work with the transitions seen in BPTI from DEShaw Research (Science, 2010) using their long-running 1 ms trajectory as the basis for states and transitions analyzed from the Anton production trajectory. Our algorithm works by resampling snapshot conformations known to be right before transition events, creating an ensemble set of transitions preconditioned on sampling in the space near to the transitions. This enables us to explore the reduced degrees of freedom that drive the transitions and to examine the statistical foundations of the description for state transitions in BPTI. To achieve these goals we use an adaptive framework for resampling built around a parallel relational database system and with scripts controlling molecular dynamics codes running on XSEDE sponsored national supercomputers. In addition to BPTI, we will show results that start from other trajectories defined from peptides and from other long-running protein simulations. Our scripts for the initial analysis and the resampling thus readily generalize to both long and short trajectory runs and can be used to increase sampling on a broad range of transition events. 1905-Pos Board B42 Characterizing Dynamics of Anion/Pi Interactions through Molecular Dynamics Simulations Karan Kapoor1, Michael Duff2, Robert Hinde3, Jerome Baudry1, Elizabeth Howell2. 1 Center for Molecular Biophysics, University of Tennessee/Oak Ridge National Lab, Knoxville, TN, USA, 2Department of Biochemistry Cellular and Molecular Biology, University of Tennessee, Knoxville, TN, USA, 3 Department of Chemistry, University of Tennessee, Knoxville, TN, USA. Proteins are not static structures, but undergo dynamical variations at room temperature that can lead to changes in the number and strength of different non-covalent interactions. The negatively charged (Asp/Glu) and aromatic (Phe/Trp/Tyr) amino acid groups in proteins can form anion/pi interactions, that have been shown to have a distance and angle dependency. This study characterizes the dynamic stability of these interactions in RmlC (PDB1EP0), a homodimeric epimerase showing a cluster of six anion/pi pairs in crystal structure, as a function of time using Molecular Dynamics (MD) simulations. The dynamic variations found in these interactions correspond to a potential of mean force (PMF) that exhibits a relatively wide range of distances ˚ ) and angles (0 to~45deg) sampled by these anion/pi pairs. The (~4.5 to~8.5A corresponding PMF indicates an associated free energy change that can be more stabilizing (up to 5kcal/mol) than the ab initio-calculated interaction energy for the pairs in the crystal structure (2.2kcal/mol). Possible anion/pi pairs and triplets (anion/anion/pi or anion/pi/pi) not seen in the crystal structure are also observed in other conformations sampled by the protein- both at the dimer interface and active site of the protein, forming an extensive network of anion/ pi interactions covering most of the protein structure. Initial site-directed mutagenesis (SDM) experiments targeting one of these pairs, Asp84-Phe112, show that the single Phe/Met mutation decreases the stability of the protein compared to the wild type. Together these results suggest that anion-pi interactions can play an important role in maintaining both the structural stability and function of the proteins. 1906-Pos Board B43 Simultaneous Identification, Visualization, and Comparison of Complex Events in Molecular Dynamics Simulations Michael V. LeVine1, George Khelashvili1, Harel Weinstein1,2. 1 Physiology and Biophysics, Weill Cornell Medical College of Cornell University, New York, NY, USA, 2HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute of Computational Biomedicine, Weill Cornell Medical College of Cornell University, New York, NY, USA. With the ability to perform all-atoms Molecular Dynamics (MD) simulations of complex biological systems on the micro- and even millisecond timescales, the need to extract the interesting features of the molecular behavior inherent in the resulting trajectories has become more pressing. For the large molecules, short simulations on the order of nanoseconds are often considered to be metastable and quasi-harmonic, representing small fluctuations around a 380a Tuesday, February 10, 2014 single local minimum in the free energy landscape; longer simulations, on the order of microseconds, must be treated as non-equilibrium trajectories, as they often include large, anharmonic transitions between more than one minima that can lead to significant changes in the protein topology. Because of their complexity, these long simulations are generally subjected to extensive, detailed analysis of many parameters (distances, angles, etc.), often causing the interesting dynamics to be lost in a sea of minutiae. We present a new analysis method that can be used to simultaneously identify, visualize, and compare complex events in MD trajectories of proteins. The statistical approach uses sliding window principal component analysis (sw-PCA) to identify collective motions that are large but transient, which is followed by projection techniques to compare motions between trajectories. We illustrate the method by analyzing microsecond MD simulations of the bacterial leucine transporter LeuT in complex with the substrates leucine, valine, and alanine that have been shown to produce different transport phenotypes. In all three systems we identified transient, hundred nanosecond time-scale collective motions in the intracellular domains, and found that these motions were coupled to different, substrate-specific, conformational changes in the primary substrate site. Our results indicate that the method can be a powerful tool in the analysis of all-atoms MD simulations as both system size and trajectory length increase. 1907-Pos Board B44 Unraveling the Dynamics of the EF1 Hand Upon Ca2D Binding in Neurocalcin Delta Yang Yang1, Anuradha Krishnan2, Jeffrey Viviano2, Venkat Venkataraman2. 1 Department of Chemistry and Biochemistry, Rowan University, Glassboro, NJ, USA, 2Department of Cell Biology, Graduate School of Biomedical Sciences, School of Osteopathic Medicine, Rowan University, Stratford, NJ, USA. Neuronal calcium sensor (NCS) proteins comprise a family of related proteins that mediate signal transduction in response to calcium, primarily in neurons. Neurocalcin delta (NCALD) is a member of this family. Though NCALD, like other NCS proteins, has 4 EF-hand motifs that could potentially bind calcium, it has been demonstrated that the first EF hand, EF1, does not bind calcium. NCALD has been purified from the brain and the retina and is thought to play critical roles in signaling. However, the details of the calciumdependent dynamics remain to be elucidated, despite the availability of its crystal structure. To probe the calcium-dependent changes, we carried out both experimental and computational analyses. The experimental investigations comprised of analyses of local conformational changes through tryptophan fluorescence, global conformational changes through mobility on native gels, and dimerization state through gel filtration chromatography. The wild-type as well as different mutant versions of the protein were used in these analyses. Meanwhile, atomistic molecular dynamics (MD) simulations with the explicit solvent model were performed to explore the structure and dynamics of the NCALD monomer with and without Ca2þ ions. In this report, the joint results are presented to provide evidence to support a major role for EF1 hand (which does not bind calcium) in determining the response of the protein to calcium. 1908-Pos Board B45 Mechanical Properties of DNA Binding Proteins: Tales in Silico Yuichi Togashi, Naoya Tochio. Research Center for the Mathematics on Chromatin Live Dynamics (RcMcD), Hiroshima University, Higashi-Hiroshima, Japan. A variety of proteins work as molecular machines; their motion is coupled with the function, hence their mechanical properties are important. In the nucleus, many of them interact with DNA. Their mechanisms to recognize and bind to specific DNA sequences, and the underlying mechanical properties, are particularly interesting. To elucidate such mechanisms, we started with a small example, namely transcription activator-like effector (TALE), and studied its structural properties by molecular dynamics. TALE is a protein in helical shape. Its crystallographic structures with and without DNA bound suggest that it shrinks and wraps a double-strand DNA. It consists of 34 amino-acid long repeats; each repeat has a nucleotide recognition site called RVD, so that the entire repeats can specifically bind to a DNA sequence. It is widely used for TALE nucleases (TALEN) in gene editing, and some improved mutants are known. Interestingly, non-RVD mutations, which do not directly contact with DNA, can enhance the activity of TALEN (e.g. Platinum TALEN by Sakuma et al., Sci. Rep. 2013). Recent coarse-grained studies showed that TALE is soft and can be stretched much to the direction of the helical axis (Flechsig, PLOS ONE 2014). These results implies that not only the DNA binding sites but also structural and mechanical properties of the rest of the molecule may determine the performance. We investigated such properties by all-atom molecular dynamics simulations. TALE without DNA, starting from the DNA-bound conformation, elongated within nanoseconds, suggesting that it is strongly compressed when bound to DNA. We conjectured that structures easy to shrink effectively bind to DNA. Some non-RVD mutants, including that used in Platinum TALEN, indeed showed shorter equilibrium distances between RVDs, which must fit the nucleotide positions. Possible mechanisms of positioning and wrapping around DNA will be also discussed. 1909-Pos Board B46 Watching Conformational Changes in Proteins by Molecular Dynamics Simulations Kresten Lindorff-Larsen. Department of Biology, University of Copenhagen, Copenhagen, Denmark. Proteins are dynamical molecules and their ability to adopt alternative conformations is central to their biological function. Examples include motions that underlie allosteric regulation or ligand binding, or protein dynamics in enzymes that can modulate the overall catalytic efficiency. Protein motions can often be described as an exchange between a dominant, ground state structure and one or more minor states. The structural and biophysical properties of these transiently and sparsely populated states are, however, difficult to study, and an atomiclevel description of those states is challenging. In an attempt to determine how well molecular dynamics simulations can capture slow, conformational changes in protein molecules we have studied two protein systems which are known to undergo conformational exchange on the millisecond timescale, and for which structural information is available for both major and minor states. Using enhanced-sampling all-atom, explicit-solvent molecular simulations, guided by structural information from X-ray crystallography and NMR, we show that current force fields and sampling methods allow us to sample experimentally-determined alternative conformations with surprisingly high accuracy. In particular, we find that we can reversible sample both the ground state and minor state, at that the simulations capture the structure of the minor states also. Our simulations enable us to calculate the conformational free energy between the two states, and comparison with experiments demonstrates a high accuracy. Our simulations provide insight into the structural and biophysical properties of transiently populated minor states, and help reinterpret previous experimental measurements. Further, our results demonstrate that, at least in the two cases we have studied, modern simulation methods enable us to examine these otherwise ‘‘invisible’’ states of proteins and describe their structural, functional and thermodynamic properties. 1910-Pos Board B47 A Hinge Migration Mechanism Unlocks the Evolution of Green-To-Red Photoconversion in GFP-Like Proteins Rebekka M. Wachter1, Hanseong Kim2, Taisong Zou3, S. Banu Ozkan3. 1 Chemistry and Biochemistry, Arizona State University, Tempe, AZ, USA, 2 Department of Biological Chemistry, University of Michigan Medical School, Ann Arbor, MI, USA, 3Physics, Arizona State University, Tempe, AZ, USA. In proteins, functional divergence involves mutations that modify structure and dynamics. Here, we provide experimental evidence for an evolutionary mechanism driven solely by long-range dynamic motions without significant backbone adjustments, catalytic group rearrangements, or changes in subunit assembly. Crystallographic structures were determined for several reconstructed ancestral GFP-like proteins, and their chain flexibility was analyzed using molecular dynamics and perturbation response scanning. The photoconversion-competent (red) phenotype appears to have arisen from a common green ancestor by migration of a knob-like anchoring region away from the active site diagonally across the beta-barrel fold. The mutational sites appear allosterically coupled to the dampening region, while providing conformational mobility to active site residues via epistasis. We propose that light-induced chromophore twisting is enhanced in a reverseprotonated subpopulation, activating internal acid-base chemistry and backbone cleavage to provide red color. Photoconversion rate measurements provide a bell-shaped curve, indicating that the reaction is controlled by the two apparent pKa values 4.5 (5 0.2) and 7.5 (5 0.2) flanking the chromophore pKa of 6.3 (5 0.1). We tentatively assign these values to the salt-bridged residues Glu222(211) and His203(193), and suggest that reverse protonation may enhance light-induced active site remodeling. In combination, the crystallographic, dynamic and kinetic data support a mechanism that utilizes light to coordinate the transient enhancement of Glu222 proton affinity and His65(63) alpha-carbon acidity, suggesting a concerted process of proton abstraction and main-chain bond scission. Dynamics-driven hinge migration may represent Tuesday, February 10, 2014 a more general platform for the evolution of novel enzyme activities. (This work was supported by NSF Grant No. MCB-0615938 to R. M. W. and NIH Grant No. U54 GM094599 to R. M. W. and S. B. O.). 1911-Pos Board B48 A Multi-Pronged Approach for Uncovering Allosteric Networks in Caspases Jeanne A. Hardy. Chemistry, University of Massachusetts Amherst, Amherst, MA, USA. Caspases, the cysteine proteases that initiate and control apoptotic cell death, are subject to allosteric regulation by a variety of modulators. Due to their role in cell death, caspases are of interest as drug targets for diseases ranging from cancer to neurodegeneration. The most significant hurdle to their therapeutic use appears to be related to the overlapping active-site specificities for small molecule inhibitors, which do not fully reflect their true in vivo specificities for protein substrates. Due to this complication, allosteric inhibition of individual caspases or particular caspase sub-functions is of great interest. Fortunately, caspases are extremely amenable to allosteric regulation, in large part due to their remarkably plastic substrate-binding grooves, which can be modulated by a number of distinct allosteric mechanisms. Our current work is to develop a global map of the allosteric networks across the family. We have discovered allosteric sites in caspase-6 and -9 that are natively regulated by zinc and elucidated the molecular mechanism of inhibition crystallographically. We have identified other allosteric sites, unique to caspase-3, -6, -7, -8 or -9 respectively, which are controlled by phosphorylation. The allosteric networks including these sites utilize distal control of the substrate-binding groove. Based on our understanding of these mechanisms of inhibition, we have engineered an allosterically handcuffed version of caspase-7 that can be unlocked by the intracellular reduction potential. Finally, using speciallydesigned nanoparticles we have delivered caspases and induced apoptosis in cancer cells. Together these findings move us closer to a full understanding of the allosteric networks controlling caspase function and therapeutically relevant allosteric control of caspases. 1912-Pos Board B49 Regulation of Kinases: 1 Billion Years of Evolution Roman Agafonov1, Chris Wilson1, Sarita Biswas2, Dorothee Kern1. 1 Biochemistry, HHMI / Brandeis University, Waltham, MA, USA, 2 Biochemistry, Brandeis University, Waltham, MA, USA. Protein phosphorylation is an essential regulatory mechanism that affects all aspect of cellular life from division and growth to aging and death. Misregulation of the signaling cascades leads to severe detrimental effects, and in humans often associated with cancer and other diseases. Phosphorylation is performed by a class of protein called kinases. Activation and deactivation of kinases is normally under tight control and is regulated via different mechanisms that are incredibly complex. In this work we combine phylogenetic resurrection techniques with biophysical and chemical approaches to analyze the regulatory mechanisms of modern tyrosine oncokinases Src and Abl, their common ancestor and the common ancestors between several other families of tyrosin kinases. Our results show how the regulatory elements appeared and developed throughout the evolution enabling selective regulation of complex modern cascades. Membrane Protein Interactions 1913-Pos Board B50 Interactions of Dok7 with Model Membranes Containing Anionic Lipids and Phosophoinositides Amanda Buyan, Antreas C. Kalli, Mark S.P. Sansom. University of Oxford, Oxford, United Kingdom. The receptor tyrosine kinases (RTKs) are a major class of transmembrane receptors responsible for regulation of many biological processes, including development and maintenance of synapses. Many RTKs act in concert with intracellular proteins which are thought to interact with cell membranes. Downstream-ofKinase 7 (Dok7) is a soluble protein involved in the Muscle-Specific Kinase (MuSK) signalling pathway. Dok7 binds to MuSK to facilitate clustering of acetylcholine receptors at synapses. Mutations in Dok7 have been known to result in varying degrees of severity of congential myasthenic syndromes, which is characterised by impaired muscle contractions. The structure of Dok7 reveals it to contain a pleckstrin-homology (PH) and a phosphotyrosine-binding (PTB) domain. Although there is evidence to suggest how Dok7 interacts with MuSK, it is unknown exactly how it interacts with the cell membrane. Here we apply a multi-scale Molecular Dynamics simulations method to characterise Dok7’s interactions with membranes of varying lipid composition. Coarse-grained (CG) simulations are used to characterise the mechanism of 381a which Dok7 binds to the bilayer, while atomistic simulations are used to refine its interactions with specific lipids present in the bilayer. These computational studies reveal the role of phosphatidylinositol phosphates (PIP) in the interaction of Dok7 with complex cell membranes. 1914-Pos Board B51 The Difference in Arl2 and Arl3 Membrane Binding and Localization Shobhna Kapoor1,2, Simone Mo¨bitz1, Shehab A. Ismail3,4, Eyad Kalawy Fansa3, Alfred Wittinghofer3, Roland Winter1, Katrin Weise1. 1 Physical Chemistry I - Biophysical Chemistry, TU Dortmund University, Dortmund, Germany, 2Department of Chemical Biology, Max Planck Institute of Molecular Physiology, Dortmund, Germany, 3Structural Biology Group, Max Planck Institute of Molecular Physiology, Dortmund, Germany, 4 Structural Biology of Cilia, The Beatson Institute for Cancer Research, Glasgow, United Kingdom. ADP-ribosylation factor-like (Arl) proteins are small GTPases, with Arl2 and Arl3 being close homologues that share almost all their interacting partners. Despite all similarities, Arl2 and Arl3 have distinct biological functions: Arl3 is regarded as a ciliary protein, whereas Arl2 has been reported to be involved in tubulin folding and Ras signaling. Defective ciliary function results in a number of human diseases. So far, how are these different roles attained by the two homologue proteins is not a fully answered question. A recent study showed that the N-terminal amphipathic helix of Arl3 but not Arl2 can function as a GTP-dependent pocket opener, displacing myristoylated cargo from the lipid-binding pocket of the GDI-like solubilizing factor UNC119a/b [1]. This would imply that membrane-bound Arl3GTP is not able to bind UNC119a/b, since this helix is predicted to mediate Arl3 membrane binding and is only exposed in Arl3GTP, thus connecting the membrane binding capacity of Arl to its nucleotide status and the availability of the N-terminal helix. In the present study, the membrane binding behavior of Arl3, Arl2, and UNC119a has been investigated by surface plasmon resonance, atomic force microscopy, and infrared reflection absorption spectroscopy to gain insight into the role of the N-terminal amphipathic helix of Arl2/3 during membrane binding and its modulation by complexation with UNC119a. The data reveal a preferential localization of Arl2/3 in the liquid-disordered phase of heterogeneous model membranes. Unlike Arl3 and other Arf proteins, Arl2 binds to membranes in a nucleotide-independent manner. Finally, UNC119a selectively impedes membrane binding of Arl3GTP. Reference [1] Ismail SA, Chen YX, Miertzschke M, Vetter IR, Koerner C, Wittinghofer A (2012) Structural basis for Arl3-specific release of myristoylated ciliary cargo from UNC119. EMBO J 31: 4085-4094. 1915-Pos Board B52 Regulation of K-Ras Membrane Association: Calmodulin Versus PDEd Benjamin Sperlich1, Shobhna Kapoor2, Alexander Werkmu¨ller1, Simone Mo¨bitz1, Gunther Zimmermann2, Gemma Triola2, Herbert Waldmann2, Roland Winter1, Katrin Weise1. 1 Physical Chemistry I – Biophysical Chemistry, TU Dortmund University, Dortmund, Germany, 2Chemical Biology, Max Planck Institute of Molecular Physiology and TU Dortmund University, Dortmund, Germany. Ras is a small GTP-binding protein and involved in a variety of cellular processes. The isoform K-Ras4B binds with its polybasic farnesylated C-terminus to membranes and can enter multiple interactions with a wide variety of effectors. PDEd and calmodulin (CaM) are known to function as potential binding partners for farnesylated Ras proteins, leading to a modulation of the dynamics of Ras membrane association. A previous study of our group showed that PDEd is not able to extract K-Ras4B from model raft membranes; instead, an effective delivery of PDEd-solubilized K-Ras4B to the plasma membrane was proposed [1]. Since CaM exhibits additional interaction sites to the G-domain of K-Ras4B as compared to PDEd and was shown not to be required for the transport of K-Ras4B to the plasma membrane, it was suggested that calmodulin dissociates K-Ras4B from membranes [2]. In the present biophysical approach, the influence of CaM on the interaction of K-Ras4B with anionic model raft membranes has been investigated by surface plasmon resonance, atomic force microscopy and fluorescence anisotropy measurements, supplemented by infrared reflection absorption spectroscopy experiments. The results suggest a repulsion of the K-Ras4B/CaM complex from the membrane. At the end, differentiation between the function of the two farnesyl-binding proteins on K-Ras4B is envisaged. References 1) Weise K, Kapoor S, Werkmu¨ller A, Mo¨bitz S, Zimmermann G, Triola G, Waldmann H, and Winter R (2012) J. Am. Chem. Soc. 134:11503-10. 2) Bhagatji P, Leventis R, Rich R, Lin C-J, and Silvius JR (2010) Biophys. J. 99:3327-35. 382a Tuesday, February 10, 2014 1916-Pos Board B53 Role of FisB-Cardiolipin Interactions in Membrane Fission during Sporulation in Bacillus Subtilis Martha Braun1, Christopher Daniel Rodrigues2, David Rudner2, Erdem Karatekin1. 1 Cellular and Molecular Physiology, Yale University, West Haven, CT, USA, 2 Microbiology and Immunobiology, Harvard, Boston, MA, USA. Membrane fission is a fundamental process required for endocytosis, membrane trafficking, enveloped virus budding, phagocytosis, cell division and sporulation. Despite the diversity of fission reactions, there are only two fission machineries known in eukaryotes (dynamin and ESCRT-III), and none in bacteria. We describe FisB, a 254 amino acid protein which is conserved among spore-forming bacteria. FisB mediates membrane fission during sporulation in B. subtilis. Upon starvation, B. subtilis divides asymmetrically, producing a large mother cell and a small forespore. The mother cell then engulfs the forespore. Like in phagocytosis or endocytosis, engulfment ends with a fission event that releases the forespore into the mother cell cytoplasm. FisB possesses a short cytoplasmic N-terminus, one predicted transmembrane domain, and a large extracytoplasmic portion. In FisB knock-out cells, engulfment proceeds normally, but the fission step is severely impaired (Doan et al., Genes & Dev. 2013). The goal of our research is to understand how FisB mediates membrane fission. Therefore we investigate how FisB is able to form oligomers that translocate to the fission site and sever membranes. The extracytoplasmic domain (ECD) of FisB binds cardiolipin (CL) in a floatation assay and binding is highest at low salt and undetectable at 500 mM NaCl, suggesting electrostatic interactions between FisB and CL. FisB(173-220) was identified as the cardiolipin binding domain. FisB reconstituted into artificial liposomes efficiently mediates membrane mixing only in the presence of CL. Fluorescently tagged ECD binds to cardiolipincontaining giant unilammellar vesicles and induces aggregation, membrane deformation and collapse. CL is enriched at the poles of B. subtilis, presumably in microdomains that prefer negatively curved regions. We hypothesize that CL may regulate recruitment and oligomerization of FisB. 1917-Pos Board B54 Interaction of Model Lipid Vesicles with Alveolar Macrophages Robinah Maasa. Indiana University-Purdue University Indianapolis, Indianapolis, IN, USA. Macrophages play key roles in host defense by recognizing and engulfing foreign and apoptotic bodies. To accomplish this task, they rely on complex molecular interactions involving both lipids and proteins. Previous studies have shown that surface exposure of phosphatidylserine by apoptotic cells is required for their successful clearance, suggesting specific lipid-protein interactions at least for the initiation of phagocytosis of apoptotic cells. However, macrophages can engulf foreign and apoptotic bodies that substantially vary in size suggesting that non-specific interactions over a range of length scales may be relevant. Here we investigate the correlation between physical properties of lipid bilayers and their engulfment by macrophages. We use a combination of scattering and spectroscopic methods to quantify lipid interactions and flow cytometry to measure engulfment rates. Our previous engulfment measurements at 1 hour after incubation have shown preference for phosphatidylserine-rich lipid vesicles over phosphatidylcholine. However, the extent of engulfment could depend on incubation time and on the exact state of macrophages. In the current study, we measure the engulfment of lipid vesicles made of either PS or PC lipids as a function of incubation time. This important aspect is relevant to the dynamics of macrophage activity. 1918-Pos Board B55 Investigation of the Structure of Dimers of the Voltage-Gated Proton Channel Adam C. Chamberlin1, Sergei Noskov1, Feng Qiu2, Peter Larsson2. 1 Department of Biological Sciences, University of Calgary, Calgary, AB, Canada, 2Department of Physiology and Biophysics, University of Miami, Miami, FL, USA. Voltage-gated proton channels (Hv1) can operate functionally as monomers; however in vivo they are found as dimers which exhibit cooperative gating. The mechanism by which the cooperativity is enforced however is incompletely understood. However, it is known that removal of the tail region of the dimers, residues 234 to 255 in human Hv1, remove the cooperativity. Our investigation of the structure of the dimers and mechanism by which cooperativity in gating was enforced involved the extension of a homology model for the monomer which has been validated and reported in the literature, Chamberlin et al [1], to include a homology model of the dimer tail region based on the crystal structure of Fujiwara et al [2]. Subsequent investigation of the tail region of the closed and open state dimers were sampled using coarsegrained methods and further refined using all-atom molecular dynamics simulations. Finally, the gating path was sampled using targeted molecular dynamics simulations. [1] Chamberlin A, Qiu F, Rebolledo S, Wang YB, Noskov SY, Larsson HP. Hydrophobic plug functions as a gate in voltage-gated proton channels. P Natl Acad Sci. 2014;111:E273-E82. [2] Fujiwara Y, Kurokawa T, Takeshita K, Kobayashi M, Okochi Y, Nakagawa A, et al. The cytoplasmic coiled-coil mediates cooperative gating temperature sensitivity in the voltage-gated Hþ channel Hv1. Nature Communications. 2012;3. 1919-Pos Board B56 Activation of the Ca2D-Activated Chloride Channel TMEM16A Novandy K. Lim, Janine D. Brunner, Stephan Schenck, Raimund Dutzler. Biochemistry Institute, University of Zurich, Zurich, Switzerland. TMEM16A is a Ca2þ-activated chloride channel that is involved in various physiological processes. The functional behavior of the murine ion channel mTMEM16A has been characterized by electrophysiology. mTMEM16A forms anion-selective channels that are activated by Ca2þ-binding from the intracellular side. Channel activation by Ca2þ was shown to be voltagedependent with an increase of the EC50 at negative potentials. Although studies have identified residues contributing to Ca2þ binding, a mechanistic understanding of channel activation and the relationship to the voltage-dependence remained unknown. To address this question, we have determined the structure of nhTMEM16, a fungal member of TMEM16 family, which functions as lipid scramblase. Due to the close relationships within the family, however, this protein also provides a valuable framework to investigate structure-function relationships in mTMEM16A. The structure of nhTMEM16 reveals a highly conserved Ca2þ-binding site within each subunit of the dimeric protein. In this site, up to two Ca2þ ions are coordinated by six residues, five of which carry a negative charge. The location of the site within the transmembrane region provides an explanation for the voltage-dependence of Ca2þ activation observed in TMEM16A. To probe the importance of the corresponding residues, single mutants of TMEM16A were expressed in HEK293T cells and studied by excised patch electrophysiology. The Ca2þ dose-response relationships of all mutants show a shift to higher Ca2þ concentrations, indicating that the interaction of these residues with Ca2þ is key to channel activation. The structure also hints at a region in the protein, termed the ‘subunit cavity’, as a potential site of ion permeation. The proximity of the ‘cavity’ to the Ca2þ-binding site and the presence of residues previously shown to affect ion selectivity of the channel, suggests this region as the likely candidate for the ion conduction path in TMEM16 channels. 1920-Pos Board B57 Investigating the Effect of PKA Phosphorylation on Intramolecular Interactions in Purified Full Length Wildtype CFTR Stephanie Chin1,2, Mohabir Ramjeesingh3, Paul Eckford3, Christine Bear3,4. 1 Hospital for Sick Children, Toronto, ON, Canada, 2Biochemistry, University of Toronto, Toronto, ON, Canada, 3Molecular Structure and Function, The Hospital for Sick Children, Toronto, ON, Canada, 4Physiology, Biochemistry, University of Toronto, Toronto, ON, Canada. Cystic fibrosis transmembrane conductance regulator (CFTR) is an unique anion channel of the ATP-Binding Cassette (ABC) superfamily. CFTR consists of two nucleotide binding domains (NBDs), two membrane spanning domains (MSDs) and a regulatory (R) domain. There are also alpha-helical extensions and intracellular loops (ICLs) which couple the NBDs and MSDs via ‘‘coupling helices’’. The regulation CFTR channel activity involves protein kinase A (PKA) phosphorylation at the R domain. However, not much is known about the effect of phosphorylation on the intramolecular interactions of full length CFTR. We propose that phosphorylation modifies the interactions of full length CFTR, especially at the ICL4:NBD1 interface. Previous studies of a similar ABC transporter, BtuCD, have shown that intrinsic tryptophan fluorescence can detect urea sensitive changes in the coupling between the MSDs and NBDs of full length BtuCD. Thus, we decided monitor intrinsic tryptophan fluorescence to study the effect of phosphorylation on the purified full length Wt CFTR. Interestingly, we found that the urea sensitive changes in the intrinsic tryptophan fluorescence of full length CFTR were modified upon PKA phosphorylation. We were interested to determine whether this result was due to phosphorylation modifying the ICL4:NBD1 interface. In order to specifically study that interface, cysteine crosslinking studies were employed using a short cell permeable cysteine crosslinker on a cys-less CFTR mutant with two cysteines, V510C (NBD1) and A106C7C (ICL4), transfected in HEK293 cells. Phosphorylation induced by cAMP agonists in cells resulted Tuesday, February 10, 2014 383a in a significantly increased the crosslinking of cysteines at the ICL4:NBD1 interface compared to the inhibition of phosphorylation with adenyl cyclase inhibitors. This suggests that phosphorylation modifies the ICL4 and NBD1 interface. These studies further our understanding of the molecular mechanisms underlying phosphorylation dependent gating of CFTR. Through mutagenesis of this domain, we found by FRET microscopy that SOAR and STIM1 mutated in the CRAC domain loose its capacity to interact with Orai1. This finding suggests that the association of STIM1 to cholesterol resides in a discrete region in STIM1 and may play an important role for the subsequent STIM1-Orai1 association. 1921-Pos Board B58 Biphasic Influence of Bulk Anionic Phospholipids for PIP2 Gating of Kir2.1 Channels through Binding to Two Distinct Sites Sun-Joo Lee1, Jacob Gyore2, Sarah Heyman1, Colin G. Nichols3. 1 Washington University, St. Louis, MO, USA, 2The University of Iowa, Iowa City, IA, USA, 3Cell Biology and Physiology, Washington University, St. Louis, MO, USA. Inwardly rectifying potassium (Kir) channels regulate cell excitability and potassium homeostasis. Our recent analyses show that Kir2.1 channels have a distinct (‘Secondary’) anionic phospholipid (PL(-)) binding site, in addition to the crystallographically determined (‘Primary’) PIP2 activating site. Docking results suggest that PL(-)s can bind to either site and therefore might compete with PIP2 at the ‘Primary’ site and inhibit. To test this prediction we performed the following assays with purified human Kir2.1 channels reconstituted in liposomes. First, Kir2.1 activity was measured with a fixed PIP2 content and with increasing content of various PL(-)s. At higher PL(-) levels, inhibition was observed that correlated well with predicted affinity at the ‘Primary’ site. The ‘Secondary’ site is generated by residues K64 and K219. K64C mutant channels are insensitive to PL(-) and only weakly PIP2-activated, but high PIP2 sensitivity is regenerated by tethering of K64C to the membrane by decyl-MTS modification. Inhibition by PL(-)s was more potent in decyl modified ‘Secondary’ site single (K64C) and double (k64C/K219A) mutant channels. It’s likely that PL(-) binding at the ‘Primary’ site is augmented in these mutants as a consequence of increased effective PL(-) in the membrane as well as reduced electrostatic repulsion from the PL(-) at the ‘Secondary’ site. Finally PIP2 sensitivity was measured in the presence of increasing PL(-)s. The apparent PIP2 Kd was leftshifted at low PL(-) (as expected for the activatory effect at the ‘Secondary’ site), but shifted back to the right at higher PL(-)s, consistent with an inhibitory effect of bulk PL(-) at the ‘Primary’ site, if present at high enough levels in the membrane. Such interplay between PIP2 and other PL(-)s on Kir2.1 channel gating can be predicted by a mechanistic two-site binding model. 1924-Pos Board B61 Modeling Structure of Human Papillomavirus Type 16 E5 Protein - a Molecular Dynamics Simulation Study Dhani R. Mahato, Wolfgang B. Fischer. Institute of Biophotonics, National Yang MIng University, Taipei, Taiwan. Human papillomaviruses (HPV) infect mucosal and cutaneous epithelial cells leading to precancerous lesions. The HPV genome encodes three oncoproteins: E5, E6 and E7 from which E5 is the least understood. E5 of HPV-16, one of the ‘‘high risk’’ types of HPV strains, is an 83 amino acid hydrophobic membrane protein, with three hydrophobic transmembrane domains (TMDs). It oligomerizes into dimers or higher oligomers which form ion channels most likely by forming hexameric bundles. Computational modeling is used to obtain structural and functional features of this protein. The three TMDs of E5 are identified using secondary structure prediction programs. The TMDs are assembled into a monomer by a ‘Sequential’ and ‘Simultaneous’ docking approach in which the conformational space of the three helices is screened by simultaneously altering distance, tilt and rotational angle between them. In a consequent step, loops linking the three helices are added using the program Loopy. Finally six monomers are assembled into a hexameric bundle. The bundle with TMD2 lining the pore remains intact allowing formation water filled pocket during entire 100 ns MD simulations. The water pocket formed by the six TMD2s of the bundle is mostly mantled by hydrophilic residues such as Ser-35, 37 and Thr-38, 40. Bundles with the other two TMDs, TMD1 and TMD3, mantling the pore are energetically almost undistiguishable from the bundle with TMD2 facing the pore. With TMD2s facing the pore, ion channel activity possible. All asymmetric bundle architectures account for interactions of E5 with host proteins. 1922-Pos Board B59 Conformational Changes that Opens TrkH Ion Channel Hanzhi Zhang, Yaping Pan, Ming Zhang. The Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, USA. A high intracellular potassium ion concentration is required for many essential cellular functions. To carry potassium ions across membranes, organisms must express potassium ion transport proteins, such as proteins in the Superfamily of Potassium Transporters (SKT). TrkH, a member of SKT, is required for bacterial growth in environments with low external potassium concentration. Previous studies showed that TrkH is an ion channel and ATP increases channel activity through an associated cytosolic protein, TrkA, which forms a homotetrameric ring. However, whether ATP regulation is preserved in TrkH of other organisms, and how ATP upregulates TrkH via TrkA are still not clear. Crystal structures of TrkH and TrkA suggest that movement of a tilted helix in TrkH and a conformational change in the TrkA tetrameric ring are required for the gating process. We have expressed and purified TrkH and TrkA from various pathogens, reconstituted them into liposomes and will examine the effects of ATP and other potential ligands on their activity. We will test our structureinspired gating model by measuring the rate of crosslinking between strategically placed pairs of cysteine mutations. 1923-Pos Board B60 Identification of a Cholesterol Recognition/Interaction Amino Acid Consensus Domain in STIM1 and its Role in SOCE Jonathan E. Pacheco, Luis Vaca. Cellular biology, Instituto de Fisiologı´a Celular, Mexico City, Mexico. Store-operated calcium entry (SOCE) is a mechanism of calcium influx activated after the depletion of intracellular stores. The main components of this mechanism are Orai1, the calcium channel, and STIM1 a calcium sensor, which oligomerizes and activates Orai channels when calcium levels drop inside the endoplasmic reticulum. The activation of Orai1 requires a series of molecular rearrangements of STIM1, which culminate in the final exposition of a domain within STIM1 known as SOAR (Stim Orai Activating Region). Specialized plasma membrane regions enriched in sphingolipids and cholesterol, have been shown to modulate SOCE also. In this work, we identified in the SOAR region a cholesterol recognition/interaction amino acid consensus (CRAC) domain, which appears to be important for the STIM1-Orai1 interaction. 1925-Pos Board B62 Bcl-xL Destabilization of Ceramide Channels: Role of the Hydrophobic Groove Kai-Ti Chang, Andriy Anishkin, Marco Colombini. University of Maryland, College Park, MD, USA. Ceramide forms channels in the mitochondrial outer membrane capable of releasing proteins that trigger the execution phase of apoptosis. Bcl-xL inhibits the formation of these channels. Previous work indicated the hydrophobic groove of Bcl-xl may be the site that binds the ceramide channel resulting in its destabilization. Single residues in the hydrophobic groove were replaced with others with different biophysical properties generally resulting in a reduction of the potency of the mutated Bcl-xL but occasionally the potency was increased. Binding of fluorescent ceramide (C11 TopFlour ceramide) to the Bcl-xL protein was also affected by the mutations in a manner correlated to the functional changes in mitochondria. These results demonstrate that the hydrophobic groove is indeed the binding site. When Bcl-xL binds to fluorescent ceramide, the fluorescence is quenched compared to ceramide dissolved in isopropanol indicating that the binding only partially protects the fluorophore from quenching by water. The quenching is greater in the mutants indicating weaker binding and greater water contact. This view is supported by molecular dynamic simulations showing more motion of the bound ceramide in the mutant Bcl-xL and thus more access to water. This hydrophobic site also binds to the BH3 domain of Bax and inhibits Bax channel formation in the mitochondrial outer membrane. Some of mutants inhibited both channels to a similar extent. However, 2 mutants acted sufficiently differently on the two channels indicating overlapping but distinct binding sites. These mutants may be useful for distinguishing between these two modes of mitochondrial outer membrane permeabilization. (Supported by NSF grant MCB-1023008) 1926-Pos Board B63 Conformation Changes of a 7TM Receptor Caused by the Sample Environment as Studied by Multidisciplinary Biophysical Methods Xiaoyan Ding1, Zhen Cao1, Bo Peng1, Anthony Watts2, Xin Zhao1. 1 East China Normal University, Shanghai, China, 2University of Oxford, Oxford, United Kingdom. Choosing an appropriate sample environment is critical in structural biology, not only for trapping functionally relevant intermediate states of a membrane protein, but also for interrogation of structure, conformation and dynamics to elucidate structure-function relationships. Functional assays and structural studies should therefore be performed in the same environment. Bacteriorhodopsin (bR), a member of the microbial rhodopsin family 7TM proteins, acting as a light-driven proton pump for light energy capture in 384a Tuesday, February 10, 2014 Halobacterium Salinarum, has been studied by multidisciplinary approaches to reveal the molecular machinery of proton transfer and photocycle. For example, in order to study the functionally related structural change in M-state intermediate, different media with a high pH value have been used to extend the lifetime of M state. However, not much attention have been paid to the conformational changes that occur in the ground state, especially around the retinal binding pocket, due to local environment. Here, the conformation changes to the bR ground state, especially around the retinal binding pocket, have been studied by solid-state NMR through chemical shifts and torsion angle measurements, combined with the light-induced kinetic and UV spectroscopies. All the experimental results have been discussed in the context of X-ray crystal structures, and possible mechanisms have been proposed. 1927-Pos Board B64 Stripping the CLC-ec1 Dimerization Interface: An Investigation into the Role of Van Der Waals Interactions in Membrane Protein Assembly Kacey Mersch, Ankita Chadda, Venkatramanan Krishnamani, Janice L. Robertson. Molecular Physiology and Biophysics, University of Iowa, Iowa City, IA, USA. What drives membrane proteins to fold and oligomerize in lipid bilayers? One possibility is that protein-protein interactions stabilize the assembled state. In membrane proteins, interfaces are typically lined by non-polar residues offering weak van der Waals interactions (VDW), yet a large network of these might confer strong stability. To investigate the role of side-chain VDW interactions on the free energy of membrane protein complex formation, we have made a library of mutations on the dimerization interface of the CLC-ec1 Cl-/Hþ antiporter - a model system that we have developed to measure equilibrium dimerization in lipid bilayers. The interface is comprised of four alpha-helices lined by non-polar residues: I - F219, I220, I223, I227; H - L194, I197, I198, I201; P - L406, I409, I410, L413; Q - I422A, L423A, I426A, I430A, I434A. We systematically mutated each helix surface to alanine (all-ALA), stripping the interface of its VDW interactions. In all cases the protein expresses, is stable and upon reconstitution in membranes, shows Cl- transport activity comparable to wild-type CLC-ec1. We ran gel-filtration chromatography over the course of one week to screen the stoichiometry of the protein in detergent. All-ALA helix I, P & Q are stable dimers, however allALA helix H showed shifts to monomer directly after purification. Furthermore, single-mutant L194A was sufficient to shift the protein to a monomer-dimer mixture. Because helix H interacts with helix P, and all-ALA helix P is still dimeric, these results suggest that dimer stability cannot be explained by VDW interactions alone, at least in detergent. We are currently measuring the change in free energy of CLC-ec1 dimerization with these subtractive mutations to quantify the role of VDW interactions in membrane protein assembly. 1928-Pos Board B65 Sarcolipin-Mediated Regulation of SERCA by Computer Simulations Alessandro Cembran1, Alysha A. Dicke2, Alfonso De Simone3, Kaustubh R. Mote2, Vitaly V. Vostrikov2, Gianluigi Veglia4. 1 Chemistry and Biochemistry, University of Minnesota Duluth, Duluth, MN, USA, 2Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN, USA, 3Life Sciences, Imperial College, London, United Kingdom, 4Biochemistry, Molecular Biology, and Biophysics; Chemistry, University of Minnesota, Minneapolis, MN, USA. The sarco-endoplasmic reticulum Ca2þ-ATPase (SERCA) is a transmembrane pump that, upon ATP hydrolysis, pumps Ca2þ against a concentration gradient from the cytosol to the sarco- or endoplasmic reticulum, thus terminating muscle contraction and priming the cell for the next excitation-contraction stimulus1. SERCA function in skeletal muscle cells is regulated by sarcolipin2,3 (SLN), a 31 amino acid transmembrane peptide that inhibits SERCA by lowering its apparent Ca2þ affinity. By combining solid-state NMR and cross-linking experiments, we characterized the structure of the SERCA/SLN complex in its natural membrane environment in both Ca2-E1:ATP and HnE2:ATP states of the catalytic cycle. In this work, we employ molecular dynamics computer simulations to investigate SERCA’s mechanism of regulation by mapping the effects of SLN binding on SERCA’s free energy landscape, sub-microsecond structural dynamics, and allosteric coupling between the transmembrane and cytoplasmic domains. Our calculations indicate that upon binding to SERCA, SLN increases its average tilt angle with respect the unbound state. The interaction between the two proteins results in a shift of the free energy basin of SERCA and in an altered coupling between its transmembrane and cytoplasmic domains, which may be responsible for the reduced activity of SERCA in its SLN-bound state. References: (1) Moller, J. V.; Olesen, C.; Winther, A. M. L.; Nissen, P. Q Rev Biophys2010, 43, 501. (2) Mascioni, A.; Karim, C.; Barany, G.; Thomas, D. D.; Veglia, G. Biochemistry-Us 2002, 41, 475. (3) Shi, L.; Cembran, A.; Gao, J.; Veglia, G. Biophys J. 2009, 96, 3648. 495, 260. 1929-Pos Board B66 Monitoring Apolipoprotein Binding to Single Lipoproteins Michel de Messieres, Abby Ng, Cornelio J. Duarte, Alan T. Remaley, Jennifer C. Lee. National Institutes of Health, Bethesda, MD, USA. Lipoproteins such as very-low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) are key players for cholesterol transport and fundamental to understanding the mechanisms of heart disease. Apolipoproteins bind to lipoproteins and their quantity and location have been proposed to be more significant indicators of heart disease than levels of good (HDL) or bad (LDL) cholesterol. We are developing a general experimental framework to study the interaction of apolipoproteins with individual lipoproteins in vitro. Specifically, we are studying apolipoprotein C-III (ApoCIII), which inhibits the breakdown of triglyceride-rich VLDL and is correlated with hypertriglyceridemia and atherosclerosis. To measure the quantity of ApoCIII on lipoproteins, we created a two-color system by labeling lipoproteins and ApoCIII with different fluorophores. Colocalization measurements were conducted via wide-field fluorescence microscopy using chambers with thickness of less than 1 mm. Lipoproteins and ApoCIII diffuse freely and are tracked simultaneously, avoiding potential artifacts from the use of surface attachments. ApoCIII binding on individual lipoproteins is quantified and exchange rates between free and bound are characterized. Of particular interest is to capture heterogeneous phenomena that are thought to occur in vivo, such as asymmetric binding distributions, which cannot be easily identified in bulk experiments. 1930-Pos Board B67 NMDA Receptor Transmembrane Domain: Structure and Mechanism of Ion Selectivity Samaneh Mesbahi1, Lea Veras1, Jon W. Johnson2, Maria Kurnikova1. 1 Carnegie Mellon University, Pittsburgh, PA, USA, 2University of Pittsburgh, Pittsburgh, PA, USA. NMDA receptors (NMDARs) are iGluR subfamilies that are activated during synaptic transmission. The voltage dependence of NMDARs differentiates them from other iGluRs: at typical neuronal resting voltages, NMDAR channels are mostly blocked by Mg2þ, but when membrane voltage is depolarized, Mg2þ block is relieved, resulting in Ca2þ influx through NMDAR at postsynaptic sites. The mechanisms, by which NMDARs select Ca2þ for permeation over all other physiological ions, while binding Mg2þ and restricting its permeation, are not well understood. Recently, partially resolved medium-resolution structures of an NMDAR tetramer were published. The structures tremendously improve our knowledge and understanding of the architecture and design of the NMDARs. Yet, one of the key structural features, namely the ion selectivity filter and parts of the pore region of the ion channel itself, are missing from these structures. This region of the protein has not been resolved also in the earlier high-resolution structure of an AMPAR, a closely related iGluR family member. Previously a successful homology model of the NMDAR TMD using one of the potassium channel family members, NaK channel as a template, was shown to have good agreements with available experimental data [Siegler et. al., Nat Neurosci 15: 406–13]. Given recently released NMDAR structures and our previously developed homology model, we are now in position to propose a high resolution NMDAR TMD model that is based on a hybrid structure. We performed extensive molecular dynamics (MD) and targeted MD simulations of our NMDAR TMD domain model in lipid bilayer and water. We demonstrate that our proposed structure is stable in simulations and has a well-formed binding site for Mg2þ and Ca2þ. 1931-Pos Board B68 Elucidation of the Channel Activities of Gramicidin a in the Presence of Ionic Liquids (ILS) using Model Cell Membranes Hyunil Ryu1,2, Hwankyu Lee3, Iwata Seigo4, Sangbaek Choi1,2, Young-Rok Kim5, Maruta Shinsaku4, Sun Min Kim2,6, Tae-Joon Jeon1,2. 1 Department of Biological Engineering, Inha Univ., Incheon, Korea, Republic of, 2Biohybrid Systems Research Center (BSRC), Inha Univ., Incheon, Korea, Republic of, 3Department of Chemical Engineering, Dankook Univ., Yongin, Korea, Republic of, 4Division of Bioinformatics, Soka Univ., Tokyo, Japan, 5Institute of Life Sciences and Resources & Department of Food Science and Biotechnology, Kyung Hee Univ., Yongin, Korea, Republic of, 6Department of Mechanical Engineering, Inha Univ., Incheon, Korea, Republic of. Ionic liquid(IL) is a salt in the liquid state below 100 C and have been considered as eco-friendly solvent that can replace organic solvent due to their unique Tuesday, February 10, 2014 properties; non-volatility and non-explosiveness. However, the safety of ILs in aquatic environment has not been fully assessed. In this work, we investigated the effects of ILs on ion channels when they are incorporated into a lipid bilayer. We chose gramicidin A (gA) as our model protein that selectively permeates cations. The ion permeability of gA varies depending on the type of ILs. In order to measure channel activities of gAs, we used two methods; fluorescence assay utilizing using stop-flow spectrometer and measurement of ion currents across lipid bilayers using a patch clamp instrument. Furthermore, we revealed that alkyl chain length of ILs and ion strength of buffer play important roles in ion permeability and confirmed how electrostatic effects due to charges on the membrane surface changed depending on ion strength of buffer using MD simulation. As a result, we should be able to design safer ILs by taking our results into account. 1932-Pos Board B69 Tug of War in Lung Surfactant Components: MiniB Dominates over Cholesterol during Lipid Domain Formation Aishik Chakraborty1, Erica Hui1, Alan J. Waring2, Prajnaparamita Dhar1. 1 Chemical Engineering, The University of Kansas, Lawrence, KS, USA, 2 Department of Medicine, Harbor-UCLA Medical Center, Torrance, CA, USA. Lung surfactants (LS), a complex mixture of lipids and proteins present in the alveolar lining of lungs, help in lowering surface tension to near zero at expiration. Deficiency of this surfactant can lead to Neonatal Respiratory Distress Syndrome in infants, while a dysfunction of LS can cause Acute Respiratory Distress Syndrome (ARDS) that affects patients irrespective of age. Successful medical intervention such as surfactant replacement therapy (SRT) requires a good understanding of surfactant composition and function. Currently there is no consensus on the composition of LS used in SRT, particularly the interactions between components making up this mixture. Our objective was to understand the interaction of cholesterol (a component whose role and even presence in SRT is highly debated) and MiniB (a synthetic protein mimic of native surfactant protein SP-B) at air-water interface. We report the alteration in lipid domain formation of films containing 1,2-dipalmitoyl- sn- glycero- 3phosphocholine (DPPC): 1- palmitoyl- 2- oleoyl- sn- glycero- 3- phosphatidylglycerol (POPG) in the ratio 7:3 under the influence of varying concentrations of MiniB and cholesterol. Fluorescence imaging under constant compression, along with analysis of domain size distributions, reveals that MiniB increases line tension between lipid domains, and prefers to stay in fluid POPG regions, making the liquid-ordered domains smaller in size. Small amounts of cholesterol prefer packed domains, stretching them into spirals during the process, lowering their line tension. In both cases, higher concentration yields more prominent consequences in terms of the stated changes. However, mixture containing both cholesterol and MiniB shows reduction in domain size with no changes in domain shape. This suggests the dominance of MiniB over cholesterol when interacting with lipid domains, which may have important effects on the performance of synthetic LS. 1933-Pos Board B70 Dynamic Measurements of Membrane Insertion Potential of Synthetic Cell Penetrating Peptide/pDNA/Ca2D Complexes Nabil A. Alhakamy1, Cory J. Berkland2, Prajna Dhar3. 1 Pharm.Chem, University of Kansas, Lawrence, KS, USA, 2Chemical & Petroleum Engineering and Pharmaceutical Chemistry, KU, University of Kansas, Lawrence, KS, USA, 3Chemical & Petroleum Engineering, University of Kansas, Lawrence, KS, USA. Noncovalent complexation of plasmid DNA (pDNA) using cell penetrating peptides (CPPs) has been less explored due to the relatively large complex size formed and the low-level gene expression. Here, condensing synthetic CPP polyplexes using CaCl2 produced small and stable complexes, which show higher level of in vitro gene expression. Anionic (i.e., POPS and POPG) or zwitterion (i.e., POPC) phospholipid monolayers at the air-water interface are used as model cell membranes to monitor the membrane insertion potential of synthetic CPPs. The insertion potential of complexes having different cationic (dTAT, H9, K9, R9, and RH9) and amphiphilic (RA9, RL9, and RW9) peptides were recorded using a Langmuir monolayer approach that records complexes adsorption to model membranes. Further, to mimic the pH of early endosome and late endosome and lysosome, phospholipid complex interactions were recorded at normal (pH 7.4) and low (pH 4.4) pH. All the complexes studied induced disruptions in phospholipid packing, which were most pronounced for the complexes having amphiphilic CPPs (i.e., RW9 and RL9). Particularly, the surface pressure of the complexes was significantly lower at normal pH when compared to acidic pH in the presence of POPC and POPS monolayers, except for RL9 and RW9 complexes. In contrast, the surface pressure of the complexes was significantly higher at normal pH 385a when compared to acidic pH in the presence of POPG monolayer. Since the late endosomes contain an abundance of PC lipids and low pH, these results may be highly relevant to understand the efficiency of endosomal escape of these complexes. Intrinsically Disordered Proteins (IDP) and Aggregates III 1934-Pos Board B71 Secondary Metal Binding to Amyloid-Beta Monomer is Insignificant under Synaptic Conditions Thomas Branch1, Mauricio Barahona2, Liming Ying3. 1 Institute of Chemical Biology, Imperial College London, London, United Kingdom, 2Department of Mathematics, Imperial College London, London, United Kingdom, 3National Heart and Lung Institute and Institute of Chemical Biology, Imperial College London, London, United Kingdom. Synaptically released Zn and Cu can reach high mM concentrations during neurotransmission. It is thought that multiple copper ions could bind to Ab monomer and mixed Zn/Cu coordinated Ab complexes may form. This could impact the Cu coordination to Ab, and therefore may be of relevance to Ab oligomerization and toxicity in the synapse. We investigated the kinetics of multiple Cu and mixed Zn/Cu binding to Ab. We found that the second order association rate constants are on the order of 108 M1s1 and 105 M1s1, for the first and second Cu binding to Ab, and 103 M1s1 for Zn binding to Ab-Cu complex, respectively. Given that the metal ion concentration decreases by more than three orders of magnitude within 1 ms, based on our simulation of metal ion release from synaptic vesicle to the cleft, we conclude that only the first Cu binding would be of significance. Our study implies that although Zn could substantially perturb Cu coordination in Ab, it has a negligible effect on the Ab-Cu complex in the synapse, due to its slow association. 1935-Pos Board B72 Gas-Phase Conformations of a Huntingtin N-Terminal Peptide Reveal Condensed-Phase Heterogeneity with and without the Presence of a PPII Helix James R. Arndt, Samaneh G. Kondalaji, Olivia Sarver, Megan M. Maurer, Arlo Parker, Justin Legleiter, Stephen J. Valentine. Chemistry, West Virginia University, Morgantown, WV, USA. Huntingtin aggregate morphology and kinetics are modulated by the presence of two flanking sequences: a seventeen-residue a-helix (Nt17) which lies N-terminal to the amyloidogenic polyglutamine region; and a polyproline PPII helix that is C-terminal to the polyglutamine region. Nt17 is responsible for aggregate nucleation, and as such, represents an intriguing target for gaining structural insight into the early stages of N-terminal huntingtin aggregation. This study examined the secondary, tertiary, and quaternary arrangement of Nt17 using ion mobility-mass spectrometry (IMS-MS) coupled with condensed-phase covalent modification and gas-phase isotopic labeling. Monomeric Nt17 adopted two gas-phase conformations, which were derived from solution structures. These structures ranged from compact globular to elongated helical. Nt17 multimers followed the same pattern, again adopting structures varying from nonspecific globule to bundled helix. Species ranging from the monomer up to the pentamer were observed. Covalent modification studies reveal threonine-3 and lysine-6 are solvent-exposed in the multimeric form. Additionally, polyproline, in a PPII helix conformation, was incubated with Nt17. Gas-phase isotopic labeling studies (hydrogen-deuterium exchange, HDX) on the two non-covalent complexes revealed nearly the same amount of deuterium uptake per Nt17 monomer in the complex, which suggests the same binding face is involved in Nt17 multimer and Nt17-Polyproline interactions. These results provide structural insight into Nt17 multimerization, and thus, the early stages of N-terminal huntingtin aggregation. 1936-Pos Board B73 Huntingtin N-Terminal Fragment Fibrils have a Rigid Amyloid Core Flanked by Non-Amyloid Domains with Increased Dynamics Cody L. Hoop, Hsiang-Kai Lin, Karunakar Kar, Ronald Wetzel, Patrick C.A. van der Wel. Dept. of Structural Biology, University of Pittsburgh, Pittsburgh, PA, USA. In Huntington’s disease (HD) and several related disorders, the primary genetic cause is the expansion of a CAG repeat in a disease-specific gene. In HD the resulting expanded polyglutamine (polyQ) segment occurs near the huntingtin protein’s N-terminus. The disease appears to reflect a gain of toxicity, with the toxic species involving a misfolded form of the mutant protein. However, the molecular details of the misfolded state remain unknown. In vivo studies have noted the presence of amyloid-like aggregates that are formed from 386a Tuesday, February 10, 2014 N-terminal fragments of the mutant huntingtin protein. The internal fibril structure has remained under debate, largely due to the difficulty to elucidate it in any detail. Enabled by magic angle spinning (MAS) solid-state (ss)NMR, we have obtained site-specific structural and motional constraints on misfolded amyloid-like fibrils for polyQ peptides of various lengths as well as N-terminal huntingtin fragments. The latter includes the first ssNMR studies of U13C,15N-labeled huntingtin exon1. Thus, we have elucidated the location and key structural features of the amyloid core. The ssNMR data reveal the configurations of the glutamines in the amyloid cores of simple polyQ and huntingtin exon1 fibrils to be very similar (despite quite different aggregation behavior). We also obtained direct insights into the non-polyQ segments and show that these ‘‘flanking domains’’ not only fall outside the amyloid core, but also retain remarkable dynamics. We characterize the latter by MAS ssNMR, using quantitative residue-specific measurements of relaxation as well as dipolar order parameters. The distribution and relative dynamics of amyloid and non-amyloid domains suggest a mechanism by which the flanking domains may allow huntingtin binding proteins to influence the stability and formation of fibrils. Moreover, these structural data further our understanding of huntingtin’s misfolding and aggregation pathways. 1937-Pos Board B74 Initiating Polyglutamine Aggregation — Computational Clarification of the Structural Details Markus S. Miettinen1, Luca Monticelli2, Praveen Nedumpully-Govindan3, Volker Knecht4, Zoya Ignatova5. 1 Fachbereich Physik, Freie Universita¨t Berlin, Berlin, Germany, 2CNRS, Lyon, France, 3Clemson University, Clemson, SC, USA, 4University of Freiburg, Freiburg, Germany, 5University of Potsdam, Potsdam, Germany. Motivation. In many neurodegenerative diseases, such as Alzheimer’s, Parkinson’s and Huntington’s, cell death is associated with protein misfolding and aggregation. In Huntington’s and eight other neurodegenerative diseases the aggregation-prone part of the disease protein is polyglutamine (polyQ). Interestingly, most toxic to cells are not the final aggregates, but some unknown structures occurring when aggregation initiates. Their small size and fleeting nature, however, have prohibited direct experimental observation. Results. We first show that existing experimental data [1] on polyQ aggregation kinetics implies that an on-pathway polyQ dimer has a characteristic lifetime of seconds. We then use this criterion to check with extensive molecular dynamics simulations the feasibility of six speculated structures to be aggregation-initiating. We find that only structures containing beta-hairpins with interdigitated sidechains fulfill the criterion; structures containing steric zippers or alpha- or beta-helices can be excluded. Discussion. Combining our findings with recent solid state NMR data that suggests steric zippers to better fit the final fibril [2], we suggest a pathway on which aggregation is initiated by interdigitated hairpins, creating a robust template on which steric zippers may fold. Implications. The relevance of our suggested pathway stem from the relatively benign effects of the final aggregates versus the high toxicity of early soluble species. Therapeutic strategies could be aimed to encourage steric-zipper-like conformers (and thus aggregate maturation) while discouraging -hairpin formation (and thus aggregate emergence). This work has been published in Ref. [3]. [1] Chen, Ferrone & Wetzel. Proc. Natl. Acad. Sci. U. S. A. 99 11884 (2002); Wetzel. Acc. Chem. Res. 39 671 (2006); Wetzel. J. Mol. Biol. 421 466 (2012). [2] Schneider, ... & Baldus. J. Mol. Biol. 412 121 (2011) [3] Miettinen, Monticelli, Nedumpully-Govindan, Knecht & Ignatova: Biophys. J. 106 1721 (2014). 1938-Pos Board B75 The Role of Structural Dynamics in Determining the Prion Strain Diversity Dominic Narang, Anup K. Srivastava, Samrat Mukhopadhyay. Centre for Protein Science, Design and Engineering, Department of Biological Sciences, Indian Institute of Science Education and Research Mohali, Mohali/SAS Nagar, India. Prion proteins exhibit alternate structural states and are associated with a number of devastating transmissible diseases. Recent discoveries have revealed the emerging functional roles of prions in a wide range of organisms. For instance, the self-perpetuating conformational change coupled with amyloid formation of a yeast prion protein, Sup35p, a translational termination factor in yeast, is responsible for novel [PSIþ] prion phenotypes in Saccharomyces cerevisiae. The 253-residue NM domain of Sup35p is an intrinsically disordered segment and is sufficient for [PSIþ] prion initiation and propagation. The NM amyloid recapitulates one of the most spectacular phenomena of prions, namely, the strain-diversity. Earlier it has been shown that two well- defined strains of [PSIþ] can be created in vitro. The molecular origin of these strains is postulated to involve diverse, yet related, conformational states and supramolecular packing of proteins within the amyloid fibrils. However, the precise structural and dynamical variations between the prion strains and their distinct physiological impacts remain elusive. To elucidate the structural origins of the prion strains, we took advantage of the fact that NM is devoid of tryptophan and created 19 single tryptophan mutants encompassing the entire length of NM. After establishing that these mutants behave similar to wildtype, we recorded a number of steady-state and dynamic fluorescence readouts that revealed the residue-specific dynamics and supramolecular packing within the amyloids responsible for different strains. The structural differences in two prion strains provided unique molecular insight into the differential binding of Hsp104 that is known to govern the strain propagation. The strain-diversity was further elucidated using time-resolved emission spectra that provided intriguing insights into the water relaxation dynamics within the amyloid architecture. Taken together, our results provide important biophysical clues in discerning the prion strain-diversity in a residue-specific manner. 1939-Pos Board B76 Amyloidogenicity of Immunoglobulin Light Chains Kathrin Andrich1,2, Ute Hegenbart3, Stefan Scho¨nland3, Erich Wanker2, Jan Bieschke1,2. 1 Biomedical Engineering, Washington University in St. Louis, St. Louis, MO, USA, 2Max-Dellbrueck-Centrum for Molecular Medicine, Berlin, Germany, 3Department of Internal Medicine V (Hematology / Amyloidosis Center-), University Hospital Heidelberg, Heidelberg, Germany. Systemic light chain amyloidosis (AL) is a rare protein aggregation disease. It usually strikes in the wake of myeloma, which affects plasma cells in the adaptive immune system. During plasma cell development, the immunoglobulin light chain (LC) genes undergo several rearrangements that leave each clone with a unique protein sequence. The produced monoclonal light chains (LC) are deposited as amyloid in AL but not in Multiple Myeloma (MM) patients. We aim to elucidate the biophysical basis of this difference. In both diseases large amounts of soluble LC are secreted into circulation and excreted with urine. Hence we hypothesize that amyloidogenicity depends on the amyloid formation propensity of the individual LC sequences rather than being a result of different LC concentrations being present in both diseases, which may also alter susceptibilities to the green tea phenol Epigallocatechin-3-gallate (EGCG). To test this hypothesis we used a simple diafiltration approach to isolate LC from AL and MM patients urine, including only cases with albuminuria less than 5% of total proteinuria. We monitored their aggregation under physiological conditions in presence and absence of EGCG over a time course of three weeks in a Thioflavin T assay and compared the aggregate sizes at different time points by semi-denaturing SDS-PAGE and filter retardation assay. We probed stabilities of native and of aggregated LC by Guanidine and thermal denaturation and imaged aggregate morphologies by atomic force microscopy (AFM). Each individual LC displayed unique characteristic aggregation kinetics. However, there were no systematic differences between proteins from MM and AL patients. EGCG treatment accelerated the formation of large aggregates that are partially stable against SDS denaturation. By determining the sequence of the LC protein via MS/MS we hope to establish a correlation between sequences, aggregation propensities and clinical parameters in AL and MM. 1940-Pos Board B77 Prediction of the Effects of the Val66Met Polymorphism and Adjacent Structured Domains on the Conformational Ensemble of an Intrinsically Disordered Protein, Brain-Derived Neurotrophic Factor Ruchi Lohia, Reza Salari, Grace Brannigan. Rutgers University, CAMDEN, NJ, USA. The discovery of Intrinsically Disordered Proteins (IDP) has challenged the structure-function paradigm and required new approaches for identifying functional mechanisms of proteins. Disease-associated Single Nucleotide Polymorphisms (SNP) are common in the disordered regions of proteins (>21.7 %), but not much is known about their effect on the conformational ensemble. Brain Derived Neurotropic Factor (BDNF) belongs to the family of neurotrophins, and facilitates neurogenesis in its short (mature) form but apoptosis in its long (pro) form. A common (4% US population) SNP that results in the Val66Met mutation in the disordered N terminus domain of the long form of BDNF (proBDNF) has been associated with various neurological and psychiatric disorders. We previously explored the effect of this SNP on likely conformations of the BDNF prodomain, using large-scale fully atomistic replica exchange molecular dynamics simulations of the disordered Tuesday, February 10, 2014 region, and found significant effects of the single point mutation on the global conformational ensemble. In the present study, we investigate the effects of the presence of the ordered region on this conformational ensemble, as well as the role of the SNP on docking of the disordered prodomain to the ordered region, using fully atomistic Hamiltonian Replica Exchange Simulations. These computational investigations complement previous NMR approaches that were restricted to the isolated prodomain, and serve as model calculations for studying the role of adjacent structured regions on conformations of intrinsically disordered regions. 1941-Pos Board B78 Structural Stability of Diabetes-Related Amylin Protofilaments: Applications to Fibril Design Ye Yuan, Bartlomiej Tywoniuk, Nicolae-Viorel Buchete. School of Physics, University College Dublin, Dublin, Ireland. We study, using atomistic molecular dynamics and coarse-grained methods, the conformational dynamics and structural stability of amyloid fibrils formed by the Islet Amyloid Polypeptide (IAPP), which is generally known as amylin. Human IAPP (hIAPP) is a co-secretion with insulin and widely found in fibril form in patients suffering with type-2 diabetes. New drugs may be developed if we understand the molecular structures of amylin fibrils, possibly leading to treatments to prevent the fibril aggregation. We build atomistic models of hIAPP amyloid protofilaments that are in agreement with previously published solid state NMR data. Our study includes different conformations and fibril topologies, and tests the effect of mutated sequences, including naturally occurring ones, that can alter the fibril stability. In particular we identify new mutations that can lead to new fibril types and compare their conformational properties. A special case is considered for amylin from human and rat organisms. In spite of relatively small sequence differences, the rodent amylin does not aggregate into fibrils, making it an excellent test case. 1942-Pos Board B79 Explosive Fibrillation Kinetics of Two-Chain Insulin Fragment Released upon Partial Digestion with Pepsin Wojciech Dzwolak1,2, Marcin Piejko1,3, Robert Dec2, Viktoria Babenko2, Agnieszka Hoang1,3, Monika Szewczyk2, Pawel Mak3. 1 Polish Academy of Sciences, Institute of High Pressure Physics, Warsaw, Poland, 2Department of Chemistry, University of Warsaw, Warsaw, Poland, 3 Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Krakow, Poland. Proteases are recognized for their role in the emergence of highly aggregationprone protein fragments in vivo. On the other hand, limited proteolysis in vitro is often used to probe different phases of amyloidogenic pathways. Here we show that moderate amounts of pepsin induce ‘‘explosive’’ fibrillation in acidified samples of bovine insulin. Biochemical analysis of the pepsininduced fibrils reveals previously unreported two-chain peptide with potent amyloidogenic properties as the main building block. The peptide (named ‘H’) comprises of N-terminal fragments of insulin A- and B-chains linked by disulfide bond between Cys7A-Cys7B and conceals up to 8 additional pepsin-cleavage sites which become protected upon fast fibrillation unless concentration of the enzyme is increased leading to complete digestion of insulin. Fibrils built of H-peptides are similar in terms of morphology (as probed by AFM) and infrared features to typical bovine insulin fibrils, but they appear to lack the ability to seed fibrillation of intact insulin. Controlled reassociation of these fragments leads to ‘explosive’ fibrillation only under non-reducing conditions implying the key role of the disulfide bonds in the amyloidogenicity of H-peptides. Our study highlights the role of dynamics of the disulfide-bonded N-terminal fragments of A- and B-chains in insulin amyloidogenesis. 1943-Pos Board B80 Elucidating the Role of Oligomers in Insulin Aggregation using Biophysical Methods Matthew T. Mawhinney, Brigita Urbanc. Physics, Drexel University, Philadelphia, PA, USA. Protein misfolding and aberrant fibrillization underlie many neurodegenerative conditions, such as Alzheimer’s and Parkinson’s disease. Insulin, which is composed of two covalently bonded peptide chains, exists in vivo mostly in a native hexameric state but becomes amyloidogenic under certain conditions: at high temperature with neutral pH (7.4) and agitation or with low pH (1.6) and quiescence. To investigate the mechanisms that drive insulin aggregation, we monitor its self-assembly into fibrils by kinetic fluorescence spectroscopy, which uses Thioflavin T (ThT), a fluorescent dye that binds to the cross-b structure of amyloid fibrils. At low pH, insulin behaves similarly to other amyloid proteins; kinetic rate of fibrillization increases with con- 387a centration. At neutral pH, we observe an increase of the kinetic rate of fibrillization with low insulin concentration (2.5 – 25 mM), whereas at higher concentrations (25 – 100 mM) the opposite trend is observed. To explain this observation, we utilize photo induced cross-linking of unmodified proteins (PICUP) and Sodium Dodecyl Sulfate-Polyacrylamide gel electrophoresis (SDS-PAGE) to determine the oligomeric population of pre-fibrillar stages of insulin self-assembly. Preliminary results show a shift toward larger oligomers at insulin concentrations in the vicinity of 25 mM. As self-assembly advances and fibrils start to form (as observed by ThT fluorescence), PICUP/ SDS-PAGE shows progressively decreased oligomer abundances. Insulin aggregation is also monitored via atomic force microscopy (AFM) to investigate differences in morphology between the two methods used to induce aggregation and the corresponding time evolution of oligomeric species. Our results are consistent with oligomer formation that is on the pathway to fibril formation, thereby elucidating a key interplay between oligomers and fibrils in insulin aggregation. 1944-Pos Board B81 DMSO Induced Breaking up of Insulin Fibrils Monitored by Vibrational Circular Dichroism Ge Zhang1, Viktoria Babenko2, Wojciech Dzwolak2, Timothy A. Keiderling1. 1 U of Illinois at Chicago, Chicago, IL, USA, 2University of Warsaw, Pasteura, Poland. Bovine insulin can form stable b-sheet-rich amyloid aggregations composing of several protofibrils and adopt variable morphologies based on the fibrillation condition. Vibrational circular dichroism (VCD) was reported as a very useful probe for charactering the chirality of amyloid aggregates and detecting formation of extended, twisted fibrils.(1) We studied the effect of adding dimethyl sulfoxide (DMSO) to aqueous insulin fibrils and monitored their destabilization by VCD. We compared two types of insulin fibrils depending on sample preparation protocol, one type can have oppositely signed induced circular dichroism for amyloid-bound THT,(2) and the other type has oppositely signed VCD.(1) Transmission electron microscopy (TEM) was used to correlate the morphology with VCD spectrum to show both the molecular morphology and supramolecular chirality aspects of the DMSO induced insulin fibril breaking-up process. The two types of insulin fibrils behaved differently on addition of DMSO, but both of them were eventually denatured by high concentrated DMSO. 1. Kurouski, D., Dukor, R. K., Lu, X. F., Nafie, L. A. and Lednev, I. K. (2012) Normal and reversed supramolecular chirality of insulin fibrils probed by vibrational circular dichroism at the protofilament level of fibril structure. Biophys. J. 103, 522-531. 2. Loksztejn, A. and Dzwolak, W. (2008) Chiral bifurcation in aggregating insulin: an induced circular dichroism study. J. Mol. Biol. 379, 9-16. 1945-Pos Board B82 The Intrinsically Disordered Termini of zDHHC S-Palmitoyltransferases Facilitate Multiple Regulatory Functions Krishna D. Reddy1, Jeremy D. Baker1, Bin Xue2, Robert J. Deschenes1, Vladimir N. Uversky1. 1 Molecular Medicine, University of South Florida, Tampa, FL, USA, 2Cell Biology, Microbiology, and Molecular Biology, University of South Florida, Tampa, FL, USA. zDHHC protein acyltransferases (PATs) are a family of membrane proteins that catalyze the reversible post-translational lipidation known as palmitoylation, a process essential to normal cellular function through facilitation of membrane attachment, subcellular trafficking, and protein stability. While transmembrane proteins such as PATs are mostly ordered due to the hydrophobic membrane environment, they have cytoplasmic tails which tend to lack stable three-dimensional structure. The aim of this study was to understand the structural and functional implications of disordered PAT termini using computational, biochemical, and biophysical approaches. Intrinsic disorder prediction indicates that a conserved a-helical molecular recognition feature (MoRF) exists in the C-termini of all PATs. In the human and yeast Ras PATs (zDHHC9 and Erf2, respectively), this region was found to be essential to palmitoyltransferase function in vivo and in vitro. Additional experiments suggest that the MoRF participates in previously undescribed protein-protein interactions. The disordered termini of Erf2 also facilitate multiple regulatory post-translational modifications including phosphorylation, acetylation, and ubiquitination. As PTMs and protein-protein interactions of PATs have been poorly described, elucidation of the structure-function relationships and modifications of intrinsically disordered regions in PATs potentially represents a novel paradigm of pharmacological interrogation of protein palmitoylation. 388a Tuesday, February 10, 2014 1946-Pos Board B83 Protein Disorder in Dynein Regulation by Dynactin and NudE Jing Jie, Elisar Barbar. BB, Oregon State University, Corvallis, OR, USA. Cytoplasmic dynein is a multi-subunit protein complex responsible for retrograde transport of diverse cellular cargoes along microtubules. Dynein is comprised of heavy chains responsible for motor activity, and intermediate chain (IC) and light chains for cargo attachment and regulation. Dynein light chain LC8 is conserved across species and its binding promotes dimerization and stabilization of IC but its effect on dynein regulation remains unclear. Dynein activity is regulated by other proteins such as dynactin, which is essential for most dynein activities; and NudE, which functions in targeting dynein to the kinetochores. LC8, dynactin subunit p150Glued and NudE coiled-coil domain all bind the disordered N-terminal domain of IC. Using NMR and ITC on Saccharomyces cerevisiae and Drosophila melanogaster constructs, we show that the N-terminal helix of IC is a single a-helix (SAH) instead of a dimeric coiled-coil as predicted. NMR titrations map the exact residues of IC involved in binding to both p150Glued and NudE and the accompanying changes in structure and dynamics, while ITC experiments identify the domains of IC necessary for full binding affinity. In yeast, p150 and NudE both bind the IC SAH domain, and both interactions are enhanced when LC8 is present. In Drosophila, p150 and NudE both bind to the SAH domain but p150 also binds to a second site on IC, a nascent helix separated from the SAH domain by a 4-residue linker. A dimeric IC formed by crosslinking IC chains has increased affinity to p150 but similar affinity to NudE, indicating that bivalency causes differential effects on binding regulatory proteins, and illustrating intriguing species variation in dynactin binding to IC. These studies illustrate the importance of multiple techniques to elucidate interplay of order and disorder in providing both structural and functional versatility for complicated systems. 1947-Pos Board B84 A Fuzzy DNA Binding Region in MBD2 Recruits the Histone Deacetylase Core Complex of NuRD and Modifies Kinetics of DNA Binding David C. Williams1, Megha Desai2, Gordon D. Ginder3. 1 Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA, 2Human and Molecular Genetics, Virginia Commonwealth University, Richmond, VA, USA, 3Internal Medicine, Virginia Commonwealth University, Richmond, VA, USA. The MBD2 protein recruits and assembles the Nucleosome Remodeling and Deacetylase (NuRD) complex and thereby uniquely combines binding specificity for methylated DNA with histone deacetylation and chromatin remodeling. This complex has been implicated in methylation dependent silencing of genes during development and aberrant silencing of tumor suppressor genes during carcinogenesis. We have focused on structural and biophysical analyses of MBD2 with the long-term goal of developing methods to disrupt formation of the MBD2-NuRD complex. Along these lines, we have previously characterized and determined the structures of the coiled-coil and methylcytosine binding (MBD) domains of MBD2. More recently we have characterized an intrinsically disordered region (IDR) of ~120 amino acids linking the MBD and coiled-coil. NMR chemical shift analyses, CD, and AUC show that this region, MBD2(IDR), does not adopt a regular structure in isolation or in the context of full-length protein. Yet the MBD2(IDR) stably binds three proteins that form the histone deacetylase core of NuRD. We show that the first twothirds of the MBD2IDR are necessary and sufficient to bind the histone deacetylase core while mutating two consecutive within this region is sufficient to abrogate binding to the core complex and disrupt the function of MBD2 in cells. At the same time, adding the MBD2(IDR) to the MBD2(MBD) in vitro modifies DNA binding primarily by reducing the observed off rate and increasing affinity by ~100 fold. We find that the MBD2(IDR) does not adopt a regular fold in the presence of DNA thereby functioning as a fuzzy DNA binding region. Together our studies show that the IDR of MBD2 plays a dual role both augmenting DNA binding affinity and recruiting a large portion of the NuRD complex. 1948-Pos Board B85 C/EBPb: Case Study for the Importance of Intrinsic Disorder for Protein Function Maria Miller. Macromolecular Crystallography Laboratory, National Cancer Institute at Frederick, Frederick, MD, USA. The basic region:leucine zipper (bZIP) DNA-binding protein, C/EBPbeta is a key regulator of numerous cellular processes, but can also contribute to tumorigenesis and to viral diseases. It binds to specific DNA sites as homo- or heterodimers and interacts with other transcription factors to control transcription of a number of eukaryotic genes. Importantly, C/EBPbeta induces chromatin opening at several cell-type specific enhancers. C/EBPbeta is an intrinsically repressed protein that is activated in response to growth factors. This report discusses possible mechanisms modulating the biological activities of C/EBPbeta based on results from sequence analysis, molecular modeling, X-ray crystallography and mutagenesis studies. Analysis of primary structure indicated that C/EBPbeta is natively unstructured protein, which consists of regions with potential to fold upon binding to molecular partners and regions that retain irregular conformations independently of their environment. Conformational flexibility allows for the initial auto-inhibition via intramolecular interactions, and subsequently facilitates formation of transient intermolecular interactions that regulate C/EBPbeta’s dimerization, nuclear translocation, DNA- binding and trans-activation activities in response to cellular signals. 1949-Pos Board B86 Structural and Dynamic Analysis on Disordered H4 Histone Tail by Modified AWSEM-MD Hao Wu1, Garegin Papoian2. 1 Biophysics Program, University of Maryland, College Park, MD, USA, 2 Department of Chemistry and Biochemistry, University of Maryland, College Park, MD, USA. DNA compaction in eukaryotic cells is mediated by positively charged octamers comprised of histone proteins. The latter consists of well folded core segments, that come together to form a central cylinder, and flexible tails, protruding out from cylinder’s rim. Despite being disordered, histone tails play an important role in bridging interactions between neighboring nucleosomes, regulating folding structure and dynamics of chromatin fibers. Histone tails, in turn, are mainly regulated via post-translational modifications, such as methylation and acetylation at various positions. Because of their flexibility and disordered nature, it has been difficult to investigate histone tails both computationally and experimentally. In particular, it is desirable to develop coarse-grained, yet accurate models of histone tails, such that subsequent nucleosomal and polynucleosmal simulations could be carried out within feasible times. To achieve this goal, we added new interactions to the associative memory, water mediated, structure and energy model (AWSEM-MD), which is typically used for folding of globular proteins or binding studies. We found that modified AWSEM-MD reproduces well the complex conformational ensemble of the H4 histone tail, obtained from atomistic simulations with explicit solvent. In particular, the cumulative and site-specific effects of various acetylation combinations are consistent with the all-atom results. Our proposed extension of AWSEM-MD may allow simulating intrinsically disordered proteins with high accuracy and computational efficiency. 1950-Pos Board B87 The Acetylation Landscape of the H4 Histone Tail David Winogradoff1, Ignacia Echeverria2, Garegin Papoian2. 1 Chemical Physics, University of Maryland, College Park, MD, USA, 2 Chemistry and Biochemistry, University of Maryland, College Park, MD, USA. The DNA of higher organisms wraps around histone proteins to form the basic unit of chromatin, the nucleosome. Each histone has N- and C-terminal tails that protrude outward from the nucleosomal surface, beyond the surrounding DNA. Histone tails, which are intrinsically disordered, play an important regulatory role for genetic processes, and, because of their high flexibility, they are difficult to characterize experimentally. Furthermore, histone tails exhibit a diverse array of post-translational modifications that alter their structure and dynamics, as well as their interactions with DNA and other proteins. We investigate the effects of increasing the degree of acetylation on histone tail H4 by performing extensive explicit solvent all-atom molecular dynamics simulation. We explore the conformational preferences of wild type, mono-, di-, tri-, and tetra-acetylated H4 histone tails. Our results demonstrate that the effects of acetylation on the H4 histone tail are both cumulative and site-specific. 1951-Pos Board B88 Comparing Solution Structures of Amylin and CGRP by Nanosecond Laser-Pump Spectroscopy and Atomistic Simulations Sara M. Sizemore1,2, Gu¨l H. Zerze3, Stephanie M. Cope1,2, Jeetain Mittal3, Sara M. Vaiana1,2. 1 Department of Physics, Arizona State University, Tempe, AZ, USA, 2Center for Biological Physics, Arizona State University, Tempe, AZ, USA, 3 Department of Chemical and Biomolecular Engineering, Lehigh University, Bethlehem, PA, USA. Amylin and calcitonin gene-related peptide (CGRP) are intrinsically disordered proteins, members of the calcitonin (Ct) peptide family. They are found with Tuesday, February 10, 2014 their respective receptors in different organs and carry out different functions. Amylin is involved in regulating glucose metabolism and is implicated in type II diabetes, while CGRP is a vasodilator involved in transmitting pain signals in the nervous system, and triggers migraine attacks. Amylin and CGRP share 47% sequence homology and are able to bind to each other’s receptors and activate cell response. Such cross-reactivity is attributed to their possible structural similarity. Solution state NMR experiments show that both peptides are disordered and locally sample transient helical states close to the N-terminus. While such short-range structural properties have been compared, it is not clear whether the long-range properties are affected or not. Here we combine results from experiments, probing both long- and short-range properties of the two peptides, with results from replica exchange molecular dynamics (REMD) simulations. To measure a long-range property directly comparable to simulations, we use a nanosecond laser-pump spectroscopy technique based on tryptophan triplet quenching. This allows probing both the end-to-end distance and the rate of end-to-end contact formation in IDPs, without using prosthetic dyes. Because of the short length of our peptides and the high aggregation propensity of amylin, this information cannot be obtained using other techniques such as FRET. Our data show that both the secondary structure content and the end-toend distance of the two peptides differ significantly, and that such differences are affected by electrostatic interactions. Both our experiments and REMD simulations indicate that long-range interactions (i.e. interactions between residues that are far away in the sequence), play a significant role in determining the peptide structural ensemble in solution. 1952-Pos Board B89 Primary Sequence Controls the Specificity and Affinity of a Small Molecule Binding to the Intrinsically Disordered Protein c-Myc Lisette M. Fred, Kaitlyn P. Gerhart, Bethany L. Zablotsky, Scott A. Barnett, Steven J. Metallo. Chemistry, Georgetown University, Washington, DC, USA. Intrinsically disordered proteins (IDPs) are characterized by high flexibility and low hydrophobic to charged residue ratio. The transcription factor c-Myc is an IDP deregulated in many forms of cancer. The protein undergoes coupled folding and binding with its obligate dimerization partner Max, which is also a disordered monomer, to form a basic helix-loop-helix leucine zipper (bHLHZip). A small molecule, 10058-F4, binds specifically within an 11 amino acid region of the bHLHZip of c-Myc, stabilizing the disordered monomer. The affinity determining residues of the 10058-F4 binding site on c-Myc were distinguished by mutating individual residues to alanine and subsequently measuring binding of 10058-F4. Mutation of both hydrophobic and certain hydrophilic residues attenuated binding of the small molecule to c-Myc. The affinity determining residues may affect binding through direct, energetically favorable contact with the small molecule or via a conformational influence on the IDP which favors binding. Within the proteome (SLiMSearch3), only two proteins are identified with five affinity determining residues. Six affinity determining residues are enough to specify c-Myc. A minimal set of these key residues were introduced into Max, which does not normally interact with 10058-F4. The novel protein, ModMax, binds 10058-F4. Although the alanine scan distinguished the necessary amino acids for binding, conservative mutations of some affinity determining residues demonstrated that 10058-F4 affinity is tunable. Upon substitution of Y to W, the affinity was improved by an order of magnitude. On the other hand, the affinity decreased five-fold upon substitution of E to N. Primary sequence alone, without extended secondary and tertiary structure, is sufficient to confer both specificity and affinity of a small molecule-IDP interaction. 1953-Pos Board B90 Interaction of the Intrinsically Disordered c-Myc Oncoprotein with Racemic and Enantiopure Small Molecules Kaitlyn P. Gerhart, Steven J. Metallo. Chemistry, Georgetown University, Washington, DC, USA. The prevalence of intrinsically disordered proteins (IDPs) in cell signaling and disease makes them significant targets. Despite the absence of defined tertiary structure, small molecules can bind IDPs at sites determined by a short, linear segment of the protein’s primary sequence. The oncoprotein c-Myc, a transcription factor that must undergo coupled folding and binding to its obligate partner Max in order to interact with DNA, is an ideal system for understanding specificity in small-molecule binding to IDPs. Three small molecule interaction sites exist in the bHLHZip region of c-Myc, the segment necessary for coupled folding and binding to Max. The chiral small molecule 10074-A4 interacts with one of these sites (Myc372-389). The presence of Myc372-389 induces small molecule circular dichroism of racemic 10074-A4, indicating an enantiospecific interaction. We have synthesized the pure R and S enantiomers of 10074-A4 (as well as pure enantiomers of derivatives) and found that at 10 389a uM and above the compound can undergo a transition upon addition to water from an aggregate, to a dispersed molecule, to an assembled chiral complex with a strong CD signature. SPR measurements indicate interaction between Myc and 10074-A4. These data suggest multiple possible binding modes. We also report the hydrodynamic radius of the bHLHZip region of c-Myc, as determined by fluorescence correlation spectroscopy and dynamic light scattering, under different conditions including in the presence of various small molecules and c-Myc’s obligate partner Max. 1954-Pos Board B91 The Intrinsically Disordered C-Terminal Tails of E. coli SingleStranded DNA Binding Protein Regulate Cooperative Binding to Single-Stranded DNA Alexander G. Kozlov1, Elizabeth Weiland1, Anuradha Mittal2, Vince Waldman1, Rohit V. Pappu2, Lohman M. Timothy1. 1 Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, MO, USA, 2Biomedical Engineering & Center for Biological Systems Engineering, Washington University, St. Louis, MO, USA. E. coli single strand DNA binding protein (SSB) is one of the key proteins in DNA replication, recombination and repair. SSB functions as a homotetramer and binds ssDNA in different modes using either all four subunits ((SSB)65 mode) or two subunits ((SSB)35 mode), which are regulated by salt concentration and SSB binding density. These binding modes display very different ssDNA binding properties with (SSB)35 mode showing highly cooperative binding. Each SSB subunit (177 amino acids) consists of two domains: an Nterminal DNA binding core containing an oligonucleotide/oligosaccharide binding (OB) fold (residues 1-112) and an intrinsically disordered (ID) C-terminal tail (65 residues). While the conserved last nine amino acids of the C-terminal tail (‘‘the tip’’) provide the site for interaction with more than a dozen metabolic proteins the role of the ID linker (56 amino acids) remains unclear. Here we show that the amino acid composition and length of the IDL affects the ssDNA binding mode preferences of SSB protein. Surprisingly the number of IDLs and the lengths of individual IDLs together with the acidic tip contribute to highly cooperative binding in the (SSB)35 binding mode. Atomistic simulations suggest that cooperative binding correlates with preference of IDLs for globular conformations (supported by NIH grant GM030498 (TML) and NSF MCB 1121867 (RVP)). 1955-Pos Board B92 Assessing Binding Perturbation due to Artificial Vibrational Probe Groups in the Nucleoprotein-Phosphoprotein Complex of the Nipah Virus Rebecca B. Wai1, Shana R. Burstein1, Sara K. Hess1, Jenny Erales2, Sonia Longhi2, Casey H. Londergan1. 1 Chemistry, Haverford College, Haverford, PA, USA, 2CNRS Marseille, Marseille, France. The binding interaction between the intrinsically disordered nucleoprotein tail and the phosphoprotein of the Nipah Virus (NiV) involves both disorder-toorder transition and fuzzy binding. To examine the dynamic structure and the conformational distribution of this interaction, a site-specific thiocyanate (SCN) vibrational probe was incorporated at many sites on the binding region of the NiV NTAIL. Since this binding is likely driven by hydrophobic forces, replacing a non-polar amino acid side chain with a polar probe could perturb binding. Isothermal titration calorimetry (ITC) experiments were designed to determine the extent of disruption to binding thermodynamics. The ITC results were then used to inform the interpretation of the vibrational spectroscopy data and measure the importance of single amino acids in maintaining this ‘‘fuzzy’’ binding interface. 1956-Pos Board B93 Claws, Disorder, and Conformational Dynamics of the C Terminal Region of Human Desmoplakin Charles E. McAnany, Cameron Mura. Chemistry, University of Virginia, Charlottesville, VA, USA. Cellular adhesion is governed by desmosomes, which are large inter-cellular junctions that act by tethering the intermediate filaments of separate cells. Intermediate filaments bind to a protein known as desmoplakin (DP), which in turn is linked to a membrane-bound cadherin complex. The serine-rich C-terminal region of DP was recently shown to modulate its binding to intermediate filaments. Several phosphorylation sites on the C-terminal region have been identified via mass spectrometry, and have been shown to regulate the binding strength. To elucidate the molecular mechanism of this coupling, and the role of specific post-translational modifications (PTM), we are using molecular dynamics simulations to examine the structural behavior of several forms of desmoplakin (wild-type, mutants, with and without PTMs). Our results indicate 390a Tuesday, February 10, 2014 that the phosphorylation of residue S2849 leads to the formation of an arginine claw that is absent in the non-phosphorylated protein. This finding at least partly elucidates the phenotypes stemming from several disease-linked human mutations in DP. We are currently determining if R2834H, a mutation that has been linked to arrhythmogenic right ventricular cardiomyopathy, disrupts the claw structure; we are also examining the effects of methylation of R2834, which has recently been shown to control the extent of phosphorylation. This work will illuminate the structural mechanisms by which DP adhesion is ultimately controlled. 1957-Pos Board B94 The Role of Higher-Order SPOP Oligomers for Localization to Cellular ‘‘Bodies’’ and Ubiquitination Activity Melissa R. Marzahn1, Jihun Lee1, Suresh Marada2, Amanda Nourse1, Huaying Zhao3, Peter Schuck3, Stacey K. Ogden2, Tanja Mittag1. 1 Structural Biology, St. Jude Children’s Research Hospital, Memphis, TN, USA, 2Cell and Molecular Biology, St. Jude Children’s Research Hospital, Memphis, TN, USA, 3Dynamics of Macromolecular Assembly Section, National Institute of Biomedical Imaging and Bioengineering, Bethesda, MD, USA. Light microscopically detectable, non-membrane bound cellular ‘‘bodies’’ are large protein assemblies with liquid-like properties, but the biophysical basis of their formation is unclear. Weak, multivalent protein interactions can result in higher-order complexes and can enable the formation of cellular bodies. The inherent size heterogeneity of higher-order complexes renders them difficult to characterize biophysically. As a result, their size distributions remain largely unquantified, limiting molecular insight into their biological functions. We report a novel mechanism governing cellular body formation of the Speckletype POZ protein (SPOP), which was recently identified as tumor suppressor, is a ubiquitin ligase substrate adaptor that localizes to nuclear puncta. We demonstrate that its cellular localization is dependent upon the ability of SPOP to form higher-order homo-oligomers through indefinite selfassociation, mediated by two distinct oligomerization domains. Furthermore, in vitro ubiquitination of substrates is enhanced through higher-order self-association of SPOP, suggesting that SPOP puncta are hotspots of substrate ubiquitination. One of SPOP’s domains dimerizes with nanomolar affinity yielding stable SPOP dimers as ‘‘building blocks’’ for indefinite self-association, while the other domain dimerizes with micromolar affinity, rendering SPOP oligomers highly dynamic. Together, this results in isodesmic self-association, in which each addition of a dimer occurs with the same affinity, independent of the oligomer size. From this model, we describe the size distribution of SPOP oligomers, providing for the first time a quantitative analysis of protein assemblies participating in the formation of cellular bodies. Mutations within both oligomerization domains have been observed in a variety of cancers, supporting our conclusion that SPOP self-association is important for its biological function. We propose that dynamic, higher-order protein self-association is a general mechanism underlying the formation of cellular bodies, which may serve as switches to fine-tune signaling cascades. Ribosomes and Translation 1958-Pos Board B95 Towards a Whole-Cell Model of Ribosome Biogenesis: Kinetic Modeling of SSU Assembly Tyler M. Earnest1, Ke Chen2, Jonathan Lai3, Zan Luthey-Schulten3. 1 Department of Physics, University of Illinois at Urbana-Champign, Urbana, IL, USA, 2Department of Bioengineering, University of California, San Diego, La Jolla, CA, USA, 3Department of Chemistry, University of Illinois at Urbana-Champign, Urbana, IL, USA. Ribosome biogenesis is a coordinated process involving the hierarchical association of 21 proteins to the 16S rRNA in the small subunit and 33 proteins to the 5S and 23S rRNAs in the large subunit. The process is further complicated by effects arising from the intracellular environment such as molecular crowders and the location of ribosomal operons within the cell. We report on our progress on the construction of a whole-cell model of ribosome biogenesis. Here we describe a detailed kinetic model accounting for the association of 18 of the 20 ribosomal proteins to the 16S rRNA to form the small subunit in vitro. Construction of the model is guided by the Nomura map of thermodynamic protein binding dependencies as well as kinetic cooperativity data. The complex chemical reaction network is simplified to 180 distinct assembly intermediates by removing infrequently used species. The 50 -central-3’ binding order proposed in the literature is reproduced and an alternate assembly pathway, 50 -30 -central, is predicted which accounts for 30% of the total reaction flux. Biologically relevant assembly intermediates are identified and compared to intermediates observed using cryo-electron microscopy. Integration of this assembly model into an in vivo, spatially resolved whole-cell model of biogenesis accounting for the transcription and translation of ribosomal components using realistic cellular geometry will be discussed. 1959-Pos Board B96 A Structural Model of the Ribosome-Bound Protein Insertase YidC Reveals Lateral Translocation of the Nascent Chain Abhishek Singharoy1, Stephan Wickles2, Roland Beckmann3, Klaus Schulten1. 1 Beckman Institute, Univeristy of Illinois, Urbana Champaign, IL, USA, 2 Chemistry and Biochemistry, Ludwig-Maximilian-University of Munich, Munich, Germany, 3Chemistry and Biochemistry, University of Munich, Munich, Germany. The integration of membrane proteins into the cytoplasmic membrane of bacteria usually occurs co-translationally. The universally conserved YidC protein mediates this process either individually as a membrane protein insertase, or as a membrane protein chaperone in concert with the canonical proteinconducting channel, the SecY complex. However, little is known about the structural basis of YidCs interaction with ribosome, and its co-translational insertion activity. Here, we present a structural model of YidC based on evolutionary co-variation analysis, lipid versus protein exposure and molecular dynamics simulations. The model suggests a distinct arrangement of the conserved five transmembrane domains and an amphipathic helical hairpin between TM2 and TM3 on the cytoplasmic surface of the bilayer. The model was used for docking into a cryo-electron microscopy reconstruction of a translating YidC-ribosome complex carrying the YidC substrate FOc. This structure revealed how a single copy of YidC interacts with the ribosome at the ribosomal tunnel exit and suggests a site for membrane protein insertion at the YidC protein-lipid interphase. This site was confirmed by chemical crosslinking of FOc to TM3 of YidC. Together, these data suggest a mechanism for the cotranslational mode of YidC-mediated membrane protein insertion. 1960-Pos Board B97 RNA Structural Modulation in the Heart of the Ribosome Jared J. Childs, Jirair Gevorkyan, Eda Koculi. Department of Chemistry, University of Central Florida, Orlando, FL, USA. DEAD-box RNA helicase DbpA is one of the RNA maturation factors that E. coli employs during its ribosome assembly process. DbpA binds tightly and specifically to hairpin 92 of the 23S ribosomal RNA which is located in the peptidyl transferase center. Therefore, DbpA is implicated in RNA structural rearrangement in a ribosome region that is crucial for cell survival. When the helicase inactive R331A DbpA construct is expressed in E. coli cells, a 45S particle accumulates. This particle is a misassembled intermediate of the large ribosome subunit. It is not known if the 45S misassembled particle rearranges inside the cell and forms the active 50S large ribosome subunit, or if the resulting RNA structural misfolding is so severe that the 45S particle is designated by the cell for degradation. To understand the fate of the 45S particle in the cell, the ability of the 45S particle to form a native 50S subunit is tested by pulse chase. First, in the cell expressing R331A DbpA and lacking the wild type DbpA from their genome, RNA is labeled with [5,6-3H] uridine for a specific amount of time, and then transcription of new RNA is stopped by the addition of rifampicin. Cell culture aliquots are obtained at a series of time points after stopping the transcription of new RNA, and ribosomal profile analyses are performed using sucrose gradient ultracentrifugation. The ribosome profile experiments demonstrate that the conversion of the 45S intermediate to the 50S large subunit particle does occur in the cells. The conversion rate of the 45S particle to the 50S particle is currently being measured. 1961-Pos Board B98 Simulating Ribosome Dynamics and tRNA Translocation Kien Nguyen, Paul Charles Whitford. Physics, Northeastern University, Boston, MA, USA. With advances in structure determination and continued growth in highperformance computing (HPC), molecular dynamics (MD) simulations can now be employed to study large-scale conformational rearrangements in molecular machines, such as the ribosome. In the cell, proteins are synthesized by the joint action of the ribosome and transfer RNA (tRNA) molecules, enabling messenger RNA (mRNA) to be translated into peptides. In the elongation cycle of translation, tRNA molecules and the associated mRNA move between binding sites, a process known as tRNA translocation. During translo˚ ) is coupled to large-scale collective rotacation, tRNA movement (~20-50 A tions in the ribosomal subunits. In order to better understand the physical relationship between these rotations and tRNA displacements, we use MD simulations that employ a simplified description of the energetics, which elucidate the role of sterics, and molecular flexibility during tRNA translocation. For the ribosome, we construct forcefields for which each experimentally-derived Tuesday, February 10, 2014 configuration is a potential energy minimum. Using these models, we are able to simulate spontaneous tRNA translocation events and identify robust aspects of the dynamics. We find that detailed steric interactions are a dominant contributor to tRNA translocation dynamics. These results provide a framework for understanding the interplay between structure and dynamics, and suggest strategies to experimentally modulate the physical-chemical features that govern ribosome function. 1962-Pos Board B99 Single-Molecule Profiling of Ribosome Recoding Phenomena Jin Chen, Joseph D. Puglisi. Structural Biology, Stanford University School of Medicine, Stanford, CA, USA. Messenger RNA (mRNA) sequence is central to translational control, with special sequences and secondary structures regulating translational dynamics. Shine-Dalgarno sequences, mRNA hairpins and pseudoknots, as well as nascent peptide-ribosome interactions, are known to pause or stall the ribosome. These stimulatory elements may lead to kinetic branchpoints during elongation and induce recoding events, wherein the ribosome is shunted into alternative pathways that result in either changes in reading frame or the bypassing of a region of the mRNA. Here, we present single-molecule fluorescence methods with zero-mode waveguides (ZMWs) to profile directly the translational rates of thousands of single ribosomes with codon resolution, illuminating the underlying dynamic mechanisms of recoding events. We investigated two recoding events: the 1 frameshifting in the dnaX gene and the ribosome bypassing of a 50 nucleotide untranslated region in gene60 of T4 phage. We observed multiple pathways induced by the stochastic interaction of the ribosome with the stimulatory elements; the ribosomes that undergo recoding in both frameshifting and bypassing are characterized by a pause in the rotated state. Such paused states allow unusual events in elongation and may be a central feature of translational control. 1963-Pos Board B100 Ribosome Assisted GTP Hydrolysis by EF-Tu - Mechanism and the Role of Asp21 Ram Prasad Bora. University of Southern California, Los Angeles, CA, USA. The elongation factor Tu (EF-Tu) is a member of the translational GTPase superfamily, whose GTPase activity is stimulated by the ribosome. Recently we elucidated the GTPase mechanism of EF-Tu using computer simulations (Ram Prasad et al. PNAS USA, 110, 20509, (2013)) and concluded that His84 of switch II region acts mainly in an indirect way (i. e., it neither acts as a general base nor stabilizes the TS in a major way). Additionally, we also concluded that although the proton transfer step occurs through an additional water molecule it does not constitutes the rate-limiting barrier. These computational predictions are further confirmed by a recent mutational and biochemical study (Maracci et al. PNAS USA, doi:10.1073/pnas.1412676111, (2014)). This work found that a mutation of Asp21 in the P loop also hampers the GTPase activity of EF-Tu. This observation suggested that the catalytic effect is modulated by the nature of amino acid side chain — thus we obtain a support to our proposal of allosteric control by the preorganization of the p-loop. However, in order to identify conclusively the origin of the observed mutational effects, and thereby the actual role of D21 on the GTPase activity of EF-Tu, it is essential to move to a quantitative structure function analysis rather than mere qualitative arguments. Thus, we conducted a computational study aimed at examining and quantifying the molecular origin of the catalytic effect of Asp21 on the GTPase activity of EF-Tu. 1964-Pos Board B101 Using Hydroxyl Radical Footprinting to Observe Ribosome Assembly Intermediates in vivo Ryan Hulscher. Johns Hopkins University, Baltimore, MD, USA. The assembly of the E. coli ribosome small subunit has been widely studied and characterized in vitro. Despite this, ribosome biogenesis in living cells remains poorly understood. This is a very complex process in which an rRNA is transcribed, folded, cleaved, and modified, while also binding with 20 different proteins. Very little is known about how the tertiary structure of the ribosomal RNA changes during assembly. There are a number of structure-probing methods that can be used to study rRNA in vivo, but virtually all of them 391a lack the time resolution necessary to study a process like ribosome synthesis, which is completed within a few minutes. Hydroxyl radical footprinting can be used to probe in vivo rRNA structure. The hydroxyl radicals which probe the rRNA can be produced in milliseconds using synchrotron X-rays. With this technique it is possible to examine ribosome assembly with meaningful time resolution. The hydroxyl radicals cleave the RNA backbone in solvent accessible regions, giving cleavage patterns that reflect regions of flexibility and rigidity within an RNA. For the purpose of examining ribosome assembly, it is nascent ribosomes that are of interest, not pre-existing ribosomes that are already assembled. Therefore, the nascent ribosomes must be isolated from the background of preexisting ribosomes. It has been shown that cells can take up labeled nucleosides that have been added to their growth media and incorporate them into nascent RNA transcripts. These can then be isolated using affinity methods. Once the nascent, assembling rRNA has been isolated, it can be analyzed by primer extension. The reverse transcriptase terminates at the cleavage sites. The cDNA fragments are then able to be analyzed by either slab gel, capillary electrophoresis, or high-throughput sequencing methods. 1965-Pos Board B102 Exploring the Mechanism of Dhh1-Mediated Translational Repression Aditya Radhakrishnan, Rachel Green. Johns Hopkins University School of Medicine, Baltimore, MD, USA. The yeast protein Dhh1, along with its orthologs in higher eukaryotes, have long been implicated in the regulation of protein expression by activation of mRNA decapping and subsequent degradation. More recent studies have argued that repression of protein production by Dhh1 occurs via a capindependent mechanism. Through a combination of in vitro and in vivo studies using reporter assays and protein tethering, we show that translational repression by Dhh1 occurs concurrently with the formation of mRNA species ‘‘over-loaded’’ with ribosomes - or polyribosomes. Through these studies, we are further able to establish a minimal functional unit of Dhh1 - comprising only of the two central RecA domains - which is capable of engendering general translational repression. We are currently complementing these studies with high-throughput ribosome profiling analysis to ascertain the nature of Dhh1-mediated translational control across the genome with nucleotide resolution. These experiments will allow us both to look at endogenous genes affected by deletion or overexpression of Dhh1 and to look at reporter constructs again in the presence of tethered Dhh1 protein. 1966-Pos Board B103 Extra-Coding Characteristics of hERG mRNA are Essential for Channel Function Marika L. Osterbur, Thomas V. McDonald. Molecular Pharmacology, Albert Einstein College of Medicine, Bronx, NY, USA. The KCNH2 gene encodes the hERG protein, the alpha subunit of the rapid delayed rectifying potassium channel. This potassium channel plays an essential role in cardiac repolarization, and malfunction of this channel caused by mutation leads to Long QT Syndrome, type 2 (LQT2). LQT2 causes increased repolarization time in the heart, leading to ventricular arrhythmias, syncope and sudden death. There are over 600 documented mutations in KCNH2 associated with Long QT syndrome, with more mutations being reported regularly. These mutations occur throughout the length of the gene, without definitive mutational hotspots. While much investigation has been done to characterize the impact that these mutations have on the hERG channel function, little investigation has been focused on what causes the hERG channel to be intolerant to mutation. Our hypothesis is that ‘‘extra-coding’’ characteristics on the mRNA level, such as GC content, rare codon usage and mRNA structure play a critical role in determining correct protein synthesis for the hERG channel, and that mutational changes that disrupt these characteristics lead to Long QT Syndrome. To investigate this hypothesis first, hERG SNPs will be analyzed to both identify trends in disease-causing SNPs, and to find differences between disease-causing and benign SNPs such as changes in local GC content or disruption of codon usage frequency. Secondly, using a codon-modified hERG mRNA with decreased rare codon usage, GC content and CpG islands when compared to native hERG mRNA, the role of mRNA structure and ribosomal movement in determining secondary and tertiary protein structure will be investigated. 392a Tuesday, February 10, 2014 DNA Structure and Dynamics II 1967-Pos Board B104 Targeting Human Telomeric G-Quadruplex DNA by Berberine Analogs: A Comparative Biophysical Investigation Debipreeta Bhowmik1, Gopinatha Suresh Kumar2. 1 Chemistry, Indian Institute of Chemical Biology, Kolkata, India, 2 Chemistry, Indian Institute of Chemical Biology, Kolkata, India. Nucleic acids are potential target molecules in various anticancer therapies. Understanding how drug molecules interact with nucleic acid has become an active research area at the interface between chemistry, molecular biology and medicine. Berberine is the most widely known alkaloid belonging to the protoberberine group, exhibiting myriad therapeutic applications. The anticancer potency of berberine is thought to emanate from its strong interaction with nucleic acids, and inhibition of the enzymes topoisomerases, telomerases. Berberine also binds strongly to the G-quadruplex structure, an alternative DNA structural motif. The capability of berberine analogs bearing substitution at 9 and 13-position to strongly bind G-quadruplex structure is studied for developing effective anti cancer therapeutics. Compared with berberine, these derivatives exhibit stronger binding affinity with G-quadruplex and the non cooperative binding affinity of berberine was propagated in the analogs also. The circular dichroism studies indicated that the alkaloid bound quadruplex DNA has a fold similar to the unbound form. In all cases, the stoichiometry was found to be one mole of ligand binding per mole of quadruplex. Calorimetric results indicated that the interaction of these analogs with the quadruplex was entropy driven phenomenon. The negative heat capacity changes in all systems along with significant enthalpy-entropy compensation may be correlated to the involvement of multiple weak non-covalent forces in the complexation process. The amino alkyl substitution at 9-position were found to be more effective in stabilizing G-quadruplex structure compared to the phenyl alkyl substitution at 13-position. Detailed studies on these analogs stabilizing telomeric G-quadruplex-DNA through entropy driven process with high binding affinity shall be presented that enable consideration as a leads compounds for telomerase inhibition and anticancer therapy. 1968-Pos Board B105 Studying Ligand Binding and Site-Specific Mode of DNA Binding by Gamma-Butyrolactone Receptor Protein CprB from Streptomyces Coelicolor A3(2) using Two Different Fluorescence Techniques Anwesha Biswas1, G. Naresh Patwari1, G. Krishnamoorthy2, Ruchi Anand1. 1 Department of Chemistry, Indian Institute of Technology Bombay, Mumbai, India, 2Department of Chemical Sciences, Tata Institute of Fundamental Research, Mumbai, India. Quorum sensing is a cell density dependent phenomenon that utilizes inducers like g-butyrolactones (GBLs) and their receptor proteins in Streptomyces species to control expression of a plethora of genes initiating antibiotic production and other secondary metabolic pathways. The receptor proteins regulate by binding to the DNA in the promoter regions of genes; release from the DNA takes place on binding to their specific GBL molecules, initiating the expression of the downstream genes. Several cognate GBLs binding to the GBL receptor family of proteins remain elusive. Here, using the only structurally characterised member of this family, CprB from Streptomycescoelicolor A3(2) as a model system, we suggest tryptophan quenching as a method for ligand screening, for the family. The intrinsic fluorophore tryptophan (W127) is a conserved residue in the family residing within the ligand binding pocket of CprB. Docking studies show interaction of GBLs with W127 and has been identified as the cause of fluorescence quenching observed on administration of two chemically synthesized GBLs (http://dx.doi.org/10.1021/ jp503589h) CprB is also known to specifically bind various promoter sequences. Though structural breakthrough has been achieved for the complex with a consensus sequence, there is dearth of information on the mode of binding to the others. To delineate the same, motional dynamics of 2-aminopurine (2-AP) has been monitored after its incorporation at different positions within the consensus sequence and a biologically relevant cognate sequence. Comparing the dynamics restriction of 2-AP across the two sequences has helped reveal a signature pattern of DNA binding by CprB. The study highlights how the technique can be a powerful tool to understand the mode of binding even in the absence of structural breakthrough (Manuscript under review). 1969-Pos Board B106 DNA Pseudoknots with Appropriate Loop Lengths and Sequence Complementary to the Stem form Stabilizing Base-Triplet Stacks Calliste Reiling, Irine Khutsishvili, Luis A. Marky. University of Nebraska Medical Center, Omaha, NE, USA. Pseudoknots have been found to play important roles in RNA function, such as the critical roles in altering gene expression by inducing ribosomal frameshifting in many viruses and in the 5’ UTR of mRNA as riboswitches. We used a combination of UV spectroscopy and differential scanning calorimetry to investigate the unfolding of DNA pseudoknots that mimic the formation of a local triplet helix found with RNA pseudokkots, explaining 1 ribosomal frameshifting. Specifically, we determined the unfolding thermodynamics for the following DNA set of pseudoknots with sequence: d(TCTCTTnAAAAAAAA GAGAT5TTTTTTT), where the length of the ‘‘Tn’’ loop was varied from n ¼ 5, 7, 9, and 11. The increase in loop length yielded higher TMs, 53C to 59C, and folding enthalpies ranging from 60 kcal/mol to 105 kcal/mol, resulting in a significant stabilization of the pseudoknots, G ¼ 8.5 kcal/mol to 15.9 kcal/mol. We also varied the length of the loop for two sets of control molecules: straight hairpin loops and pseudoknots in which the 5’ loop is not complementary to the stem. Their increased loop length yielded slight changes in both the TMs and folding enthalpies, consistent with a slight decrease in stability with the straight hairpin loops and a slight increase in stability with the pseudoknots. Therefore, the increase in enthalpy, ~14 kcal/mol per step of two loop thymines, is explained in terms of the formation of a single basetriplet stack. For instance, the pseudoknot with the loop of 9 thymines forms two base-triplet stacks. Supported by Grant MCB-1122029 from NSF and GAANN grant P200A120231 from the U.S. Department of Education. 1970-Pos Board B107 Sequence Dependent Plectoneme Dynamics Marco Tompitak1, Behrouz Eslami Mossallam1, Gerard Barkema2, Helmut Schiessel1. 1 Instituut-Lorentz, Leiden University, Leiden, Netherlands, 2ITF, Utrecht University, Utrecht, Netherlands. In recent years, both theoretical and experimental indicators have been gathering, showing that sequence effects on the physical properties of DNA molecules contribute nontrivially to the molecule’s behavior. Here we present the first results of a study of sequence effects on the formation and dynamics of plectonemes, the supercoiled structures produced when the DNA is put under torsional stress. Using, for the first time in this context, a fully sequencedependent, non-coursegrained rigid base pair model for the DNA molecule, we examined the process of sliding a formed plectoneme along a DNA molecule in its entirety as a mechanism for plectoneme transport. We were able to map out the relevant energy landscapes and we find that we can rule out sliding as the dominant transporation mechanism. 1971-Pos Board B108 Mismatched DNA Base Pairs Show Increased Conformational Fluctuations Adelaide Kingsland, Lutz Maibaum. Chemistry, University of Washington, Seattle, WA, USA. Base pair mismatches in DNA can have many adverse consequences, yet the exact mechanism by which mismatches are repaired is unknown. Both matched and mismatched DNA sequences were studied using molecular dynamics in biased and unbiased simulation. Significant differences were found between matched and mismatched pairs in structure, hydrogen bonding, and base flip work profiles. Mismatched pairs show greater movement perpendicular to the DNA strand and a lower free energy barrier for base flip than matched pairs. This supports experimental findings that the primary mechanism utilized by mismatch repair enzymes is to fully flip the base into the active site. 1972-Pos Board B109 The Study of Complexation Process between Cationic Gemini Surfactants and DNA using Structural and Spectroscopic Methods Weronika J. Andrzejewska1, Michalina Skupin1, Magdalena Murawska1, Andrzej Skrzypczak2, Maciej Kozak1. 1 Macromolecular Physics, Adam Mickiewicz University in Poznan, Poland, Poznan, Poland, 2Faculty of Chemical Technology, Poznan University of Technology, Poland, Poznan, Poland. Dicationic (gemini) surfactants are intensively studied group of chemical compounds, because of the broad range of applications in medicine, chemical technology or pharmaceutical industry. In solution they can form with nucleic acids the complex structures (lipoplexes), which can be used as drug delivery systems Tuesday, February 10, 2014 in nonviral transfection. Lipoplexes in gene therapy offer efficient introduction of a therapeutic material to the living cells. Gemini surfactants also allow introduction of a transgene without inducing natural immunological response and release it inside the cell. In our study, we analyzed the process of complexation of cationic gemini surfactants (3.30 - [1,6- (2,n-dioxyalcane)] bis(1-dodecyloxyimidazolium dichlorides)) with DNA, using small angle X-ray scattering, circular dichroism spectroscopy and gel electrophoresis. Surfactants which have been used had of variable length of the spacer group. We observed the formation of stable complexes in these systems and the process of complex formation was reproducible, efficient and immediate. The research was supported by research grant (UMO-2011/01/B/ST5/00846) from National Science Centre (Poland). 1973-Pos Board B110 DNA-Binding Properties of Peptide-Functionalized Graphene Quantum Dots Bedanga Sapkota1, Mirela Mustata1, Jian Zhang2, Gevorg Grigoryan2, Meni Wanunu3. 1 Department of Physics, Northeastern University, Boston, MA, USA, 2 Department of Computer Science, Dartmouth College, Hanover, NH, USA, 3 Department of Physics and Chemistry/Chemical Biology, Northeastern University, Boston, MA, USA. We present here the synthesis and characterization of nanoscale materials with DNA binding properties. We have functionalized graphene quantum dots (GQDs) with graphite-binding peptides to obtain graphene/peptide conjugates. The conjugates form stable 2D crystalline beta-sheet domains on the graphene surface. Upon incubation with DNA, we observe that DNA binding to the domain is directional, and the DNA preferentially aligns to the peptide direction. Upon binding the peptide-functionalized GQDs to DNA we observe an architectural role of GQDs in global and local flexibility, looping, and compacting of DNA. We use worm like chain (WLC) model to determine the persistence length of DNA in the absence and in the presence of GQDs, confirming a decreased persistence length upon GQD binding. Further, we find that the presence of peptide augments the number of DNA binding sites, as compared with exposure of bare GQDs to DNA. Our results show that peptide-modified GQDs can be a potentially ideal nanomaterial mimic of DNA-binding proteins similar to nucleosomal structures and other DNAmetabolic enzymes. 1974-Pos Board B111 High-Affinity Fluorescence Sensing of G-Quadruplexes D. Cibra´n Pe´rez-Gonza´lez1, Flor Rodrı´guez-Prieto2, J. Carlos Penedo1. 1 University of St Andrews, St Andrews, United Kingdom, 2Universidade de Santiago de Compostela, Santiago de Compostela, Spain. Guanine-rich DNA and RNA sequences can adopt highly polymorphic fourstranded structures, so-called G-quadruplexes (GQs), which play an important role in different biological processes such as telomere maintenance, abortive transcription and gene regulation. Their biological relevance has increased the interest in the development of probes to sense GQs and the study of these compounds as potential anticancer agents. Although a wide variety of fluorescent sensors have been reported for GQ detection so far, none is a ratiometric probe. Ratiometric probes are powerful sensors for which the relative variation between two emission peaks constitutes an absolute observable, avoiding the unwanted photobleaching and background effects. In the present work, we introduce a couple of selective sensors. A compound for ratiometric studies that undergoes excited-state intramolecular proton transfer (ESIPT) and a naturally occurring compound that lights up when bound to GQs. The interaction of both fluorophores with several GQ-forming sequences was tested using steady-state and lifetime binding assays. Single-, double- and triple-stranded DNAs were used as controls. We demonstrate that ESIPT probes can be selective and quantitative ratiometric GQ sensors, thanks to the presence of two different emission peaks with good spectral separation. Additionally, some natural probes and their derivatives can act as fluorescence light-up probes in the determination of GQs with a remarkably high affinity when compared with most of the popular sensors. Our natural compound presents most of these characteristics making it a prominent candidate to develop next generation of GQ sensors. 1975-Pos Board B112 Single Molecule Measurements of the Unfolding Behavior of Diverse DNA Hairpin Assemblies Caitlin J. Cain1, Sally Ruderman1, Catherine A. Deitrich2, Diana Seminario2, Micah J. McCauley3, Mark C. Williams3, Megan E. Nunez1. 1 Department of Chemistry, Wellesley College, Wellesley, MA, USA, 2 Department of Chemistry, Mount Holyoke College, South Hadley, MA, USA, 3Department of Physics, Northeastern University, Boston, MA, USA. 393a Kinetic and thermodynamic effects of small structural changes to the DNA duplex can be studied on a single molecule level using optical tweezers. Short DNA hairpins, which can be synthesized to contain any sequence or structure of interest, serve as our model for double stranded DNA duplexes. To facilitate pulling with optical tweezers, the hairpins are attached to long flanking DNA ‘‘handles,’’ which themselves are chemically attached to beads. The forces required to fully unfold the hairpin constructs are directly related to the thermodynamic stability of the hairpin structure, allowing us to quantify the effects of mismatches, lesions, intercalator binding, and other non-canonical structures that disturb the stability of double-stranded DNA. The first step in this project is to synthesize, purify, and characterize hairpin assemblies with specific sequences, utilizing a combination of solid-phase organic synthesis and molecular biology. Then, individual DNA hairpin molecules are stretched with optical tweezers. Changes in distance and force reveal that fully-matched WatsonCrick duplex hairpins unfold and refold neatly in discrete, concerted events, whereas mismatched and bubbled hairpins open and close with lower force and greater force variability. Although the overall stability of the structures is predicted well by mfold calculations, the single molecule studies allow measurement of the barrier to unfolding as well as the distance to the transition state. After validating the method for DNA alone, changes in the transition state and hairpin stability in the presence of DNA binding ligands can then be probed directly. 1976-Pos Board B113 Optimizing Tethered Particle Motion to Measure DNA Compaction by Protamine Matthew Woop, Robert D. Schwab, Ji Hoon Lee, Ashley R. Carter. Physics, Amherst College, Amherst, MA, USA. During mammalian spermiogenesis, there is a remarkable change in the nuclear morphology whereby the chromatin is completely remodeled. Specifically, the histone proteins that wrap the DNA into nucleosomes are removed, and the DNA is coated with a series of small (~100-amino-acid), arginine-rich protamines that dramatically compact the DNA into a series of toroids. This replacement and compaction 1) reduces the head size of the sperm to enhance their hydrodynamicity, 2) decreases the likelihood of DNA damage, and 3) removes the epigenetic markers passed on through histone modifications. Here, we directly observe the steps of toroid formation and the underlying mechanics. We use a single molecule biophysics assay where we tether a particle (1-mmdiameter bead) to the DNA and to the sample surface. During DNA folding, the end-to-end length of the DNA decreases, decreasing the motion of the particle. This tethered particle motion (TPM) assay is useful because we do not apply any external forces, allowing us to measure reversible folding events. Previously, we saw ~200 nm changes in length that suggest that the toroid forms by successive loops. Here, we optimized the assay to enhance efficiency and resolution by improving bead monodispersion, protein binding to the surface, and DNA purification. 1977-Pos Board B114 Comparing Effects of Different Transition Metal Complexes under Osmotic Stress in the B-To-Z DNA Transition Richard S. Preisler, Maimouna Cisse, Daniela Rey-Ardila, Aloise Diedrich, Kelsey Polak. Chemistry, Towson University, Towson, MD, USA. The equilibrium transition from B-DNA to Z-DNA is driven by salt cations and by neutral osmolytes. Transition metal complex cations, such as cobalthexammine and cobalttrisethylenediamine, stabilize the Z-DNA form of poly[d(G-C)] through electrostatic interactions with the DNA backbone and site-specific hydrogen bonds. These complexes can be used to measure the position of the B-Z equilibrium in the presence of osmolytes. Previous studies in our lab and Donald Rau’s lab (1) have shown that applying an osmotic stress in osmolyte solutions decreases the concentrations of complex required to form the more sparsely hydrated Z-DNA. We have found that the cobalttrisethylenediamine complex is less effective than the cobalthexammine complex in driving the transition, possibly because the three bidentate ligands in the former result in a lower conformational entropy than the six monodentate ligands in the latter. On the other hand, the binding of cobalttrisethylenediamine to Z-DNA appears to be more strongly subject to competition by hydrating water molecules, as indicated by greater sensitivity to osmotic stress (2). For example, a sucrose concentration of 4.07 osmolal decreased the transition midpoint concentration of the trisethylenediamine complex by 2.5-fold, while the midpoint concentration of the hexammine complex was decreased by only about onethird. Similar trends were also observed with monohydroxylic osmolytes such as methanol and dihydroxylic compounds such as propylene glycol. Future studies will compare the (þ) and (-) enantiomers of cobalttrisethylenediamine, in an attempt to explore stereospecific aspects of their binding to DNA. 394a Tuesday, February 10, 2014 Supported by Towson University Undergraduate Research Grants (to M. Cisse, A. Diedrich and D. Rey-Ardila) and by the Towson University Department of Chemistry. 1. Preisler et al (1995) Biochemistry 34, 14400-14407 2. Ashman et al (2011) Biophysical Society meeting poster presentation 1978-Pos Board B115 The Effects of Ionic Strength on the Hydrodynamic Properties of I-Motif Folding Robert T. Wright1, Samantha M. Reilly2, Randy M. Wadkins2, John J. Correia1. 1 Department of Biochemistry, University of Mississippi Medical Center, Jackson, MS, USA, 2Department of Chemistry and Biochemistry, University of Mississippi, University, MS, USA. Cytosine-rich nucleic acid sequences found in human DNA can adopt multiple intramolecular structures identified as i-motifs that are dependent on physiochemical solution conditions. The focus of this study is a four-stranded structure from the promoter of the human c-MYC gene. This compact, stable, and monomeric structure forms upon a decrease in pH (<5.0) causing hemiprotonation of a cytosine that results in a stable C-Cþ hydrogen bond. Sedimentation velocity experiments were performed with the analytical ultracentrifuge in order to determine the hydrodynamic properties of the folded i-motif. The sedimentation velocity experiments were carried out in buffer conditions that differed in pH (4.5-8.0), salt type (NaCl and KCl), and salt concentration (up to 400 mM). Experiments were run at different pH values in order to observe the linkage between the pKa of the Hoogsteen base pair and i-motif folding. High salt concentration was used to avoid non-ideality (primary charge effect) observed when nucleic acids sediment at low salt concentration. The data indicates that the S20,w value increases when the primary charge effect is overcome at a higher salt concentration. The experimental S20,w values are compared with values obtained by bead model simulations using SOMO as implemented in Ultrascan 3. (Supported by UMC AUC Facility.) 1979-Pos Board B116 Molecular Dynamics Investigation of Immobile DNA Four-Way Junctions Matthew R. Adendorff, Mark Bathe. Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA. Immobile four-way junctions (4WJs) are the core structural motif employed in structural nucleic acid nanotechnology to constrain interconnected duplexes. While it is well known that 36 sequence designs form immobile four-way junctions that may exist in one of two stacked conformational isomers, the molecular basis for their stability and stacking interactions remains obscure. For example, experiment indicates that several core sequence motifs exhibit a strong bias for one isomeric state, whereas both isomeric states are preferred equally by others. Molecular dynamics (MD) offers atomic-level insight into the structural and chemical basis for 4WJ interactions leading to preferential stability of one over the other isomeric state. To investigate 4WJ stability, we employed all-atom MD including explicit solvent and experimental ionic conditions to examine the dynamics of the canonical Seeman J1 4WJ that strongly prefers one of its two isomeric states, and compared it with several alternative sequence designs. Analysis of base-pair level duplex and crossover degrees of freedom reveals base stacking and pairing interactions that may impact overall junction stability. 1980-Pos Board B117 Ensemble Models of Nucleosome Arrays Constrained by Small-Angle X-Ray Scattering Steven C. Howell1, Wei Meng1, Kurt Andresen2, Agnes Mendonca3, Chongli Yuan3, Bing-Rui Zhou4, Yawen Bai4, Joseph E. Curtis5, Xiangyun Qiu1. 1 Physics, George Washington University, Washington, DC, USA, 2Physics, Gettysburg College, Gettysburg, PA, USA, 3Chemical Engineering, Purdue University, West Lafayette, IN, USA, 4Laboratory of Biochemistry and Molecular Biology, National Cancer Institute, NIH, Bethesda, MD, USA, 5 NIST Center for Neutron Research, Gaithersburg, MD, USA. Chromatin conformation and dynamics remains unsolved despite the fundamental role of the chromatin in genetic functions such as transcription, replication, and repair. At the molecular level, chromatin can be viewed as a linear array of nucleosomes, each consisting of 147 base pairs (bp) of dsDNA wrapped around a protein core and connected by 10 to 90 bp of linker dsDNA. Using small-angle x-ray scattering (SAXS), we have investigated how the conformations of model nucleosome arrays in solution are modulated by ionic condition as well as the effect of linker histone proteins. To facilitate ensemble modelling of these SAXS measurements, we have developed a DNA Monte Carlo move module that significantly extends the functionality of SASSIE, a program to study intrinsically disordered biological molecules. Our SAXS measurements of various nucleosome arrays together with the SASSIE generated models provide valuable solution structure information and clearly demonstrate the importance of linker histone in the formation of well-defined chromatin conformations. Additionally, the added capability of SASSIE provides a valuable tool for generating atomistic molecular structures of DNA and DNA-protein systems based on experimental scattering measurements. 1981-Pos Board B118 Torque Measurements during the Spontaneous Unbraiding of DNA Molecules Showed Large Fluctuations Attributable to the Formation of Stable DNA-DNA Interactions Carlos J. Martı´nez-Santiago, Mo´nica Ferna´ndez-Sierra, Edwin Quin˜ones. University of Puerto Rico, Rı´o Piedras Campus, San Juan, Puerto Rico. Magnetic tweezers dual-molecule braiding assays provide an adequate framework for studying the mechanical aspects underlying the process of winding two DNA molecules around each other. In its typical implementation however, this technique do not allow direct measurement of torque because the torque exerted by the magnetic traps (~103 pN nm) is at least one order of magnitude larger than the restoring torque of DNA (~ 102 pN nm). In previous work, our laboratory showed that enough elastic energy could be stored in a pair of braided DNA molecules to perform mechanical work by rotating a microscopic object in absence of manipulation. Here we have exploited that unique property of DNA to perform a real-time study of braided DNA dynamics. In our experiments, torsionally unconstrained lambda DNA molecules were bound to a surface by one end and to a microscopic dumbbell at the other. A magnetic tweezers apparatus was employed to stretch and braid the molecules. Upon creating the braid, the magnet was removed to trigger the spontaneous unbraiding of DNA. The rotational dynamics of the dumbbell was followed in real-time. Hydrodynamic equations were employed to relate the dynamical information of the dumbbells to torque values. Our most important finding is that the unbraiding process occurs in a discontinuous manner. The estimated magnitudes of torque were found to fluctuate between an upper (~ 102 pN nm) and a lower bound (~ 101 pN nm) value along the entire process. The magnitudes of the fluctuations were found to be independent of the amount of times that the molecules crossed each other. No different was found for braids of opposite handedness. We attributed these fluctuations to the formation/rupture of stable DNA interactions. 1982-Pos Board B119 Quantifying the Stability of Acridines to Ribosomal G-Quadruplexes Billy Nicholson1, Adam Green2, Samuel Cho3. 1 Physics, Wake Forest University, Winston Salem, NC, USA, 2Chemistry, The University of Tennessee, Knoxville, TN, USA, 3Physics, Computer Science, Wake Forest University, Winston Salem, NC, USA. G-quadruplexes are involved in fundamental regulatory processes, including those associated with cancer. Previous studies primarily focused on telomeric and oncogenic DNA G-quadruplexes, but recent studies have demonstrated that G-quadruplexes in ribosomal DNA or ribosomal RNA (rDNA or rRNA), may be an alternative target with a significant clinical potential through the inhibition of RNA polymerase I (Pol I) in ribosome biogenesis. The structures of ribosomal G-quadruplexes at atomic resolution are unknown and very little biophysical characterization has been performed on them. In our present study, we modeled putative ribosomal G-quadruplexes that were previously predicted using a bioinformatics algorithm and verified using CD spectroscopy to exist in a predominantly parallel topology. To quantify the relative stabilities of the acridine:G-quadruplex structures, we introduce two novel metrics for quantifying the relative stability of the G-quadruplex tetrads: 1) the center of mass base-to-base distance between diagonal guanines, and 2) the torsional angle between four guanines. Our relative free energy profiles show that the rDNA G-quadruplex structures with shorter loops are more stable for parallel topologies. The antiparallel topology was determined adopt a disordered configuration due to the lack of planarity after the simulation. To evaluate acridine molecules as a viable small molecule chemotherapeutic agent, we then rationally designed several acridine molecules. Each has a polyaromatic face that binds to the top-most G-tetrad and has amino or carboxyl side-chains to selectively optimize the shape and electrostatic complementarity. We then docked acridines to the rDNA G-quadruplexes and performed MD simulations. We identified several acridines with amino groups, and the nature of their specificity and relative stabilities depend on the rDNA G-quadruplex loop structures. Tuesday, February 10, 2014 1983-Pos Board B120 Molecular Identification of the Earthworm Amynthas Gracilis Patricia G. Morgante, Ana Caroline Conrado, Patricia S. Santiago. UNESP - C. E. Registro, Registro, Brazil. Earthworms have been widely used as bio-indicators of soil quality. In such works, the precise identification of the species is a ‘‘sine qua non’’ condition. The use of morphological parameters for species identification is difficult and, sometimes just impossible (e.g. identification of juveniles). Intending to investigate the potential use of Amynthas gracilis as bio-indicators of soil quality, we first decided to evaluate the molecular identification of A. gracilis by means of DNA markers. In order to reach this objective, DNA extractions were performed with specimens acquired from farms of earthworms. The standardization of the method was based on protocols described previously. PCR reactions were performed with the use of two molecular markers: 16S rDNA and COI. The chemical conditions of the reactions followed procedures used in the laboratory. The amplification cycles considered annealing temperatures of 44 C for 16S rDNA and 49 C for COI. Automatic sequencing was performed on 3500 Genetic Analyzer (Applied Biosystems) and whit BigDye version 3.1 for reactions. Until now, we were able to analyze just five specimens using the BLAST tool of GenBank. For both markers was possible to recovery matches of 100% of identity with A. gracilis sequences deposited in the databank, indicating the potential of these markers for molecular identification of this species. However, for both markers we were able to get matches of 98% and 99% of identity with other A. gracilis GenBank accessions, and in the case of 16S, 99% of identity with one access of A. diffringens. These results may suggest the presence of some intraspecific variability, and for 16S marker, the variability intraspecific and interspecific may be similar in some cases, what, if confirmed, could difficult its application for molecular identification. Further studies will be necessary to clarify these questions. 1984-Pos Board B121 Molecular Dynamics Investigations of Z[WC] DNA and the B to Z-DNA Transition Michael G. Lerner1, Alma Gracic1, Jinhee Kim1, Ashutosh Rai1, Alexander K. Seewald2, Benjamin L. Yee2. 1 Physics and Astronomy, Earlham College, Richmond, IN, USA, 2Computer Science, Earlham College, Richmond, IN, USA. Although DNA is most commonly found in the right-handed B-DNA structure, it is known that biologically active systems also contain left-handed ZII-DNA. We investigate the possibility that Z[WC]-DNA serves as an intermediate structure in the B to ZII transition. Molecular dynamics simulations indicate that Z[WC] forms stable structures with the current AMBER nucleic acid force field. Steered molecular dynamics simulations indicate that, for collective transitions of the whole strand, the B-Z[WC]-ZII pathway may have a lower freeenergy barrier than the direct B-ZII pathway. A careful combination of steered and targeted molecular dynamics, along with umbrella sampling, are used to produce potentials of mean force for both the direct and the B-Z[WC]-ZII pathways. 1985-Pos Board B122 Structure and Thermodynamics of Aegis Nucleotides P and Z in DNA Xiaoyu Wang1, Kenneth K. Sharp1, Shuichi Hoshika2, Stanislav Bellaousov3, Xiaoju Zhang3, David H. Mathews3, Steven A. Benner2, Raymond J. Peterson4, Jason D. Kahn1. 1 Chemistry and Biochemistry, Univ. Maryland College Park, College Park, MD, USA, 2FfAME, Gainesville, FL, USA, 3Biochemistry & Biophysics, Univ. of Rochester Medical Center, Rochester, NY, USA, 4Celadon Laboratories, Hyattsville, MD, USA. Artificially expanded genetic information systems (AEGIS) enhance synthetic biology and in vitro primers, probes, and sensors. DNA/RNA design requires the prediction of secondary structures including the expanded alphabet nucleotides. This requires nearest-neighbor thermodynamics analogous to those used for DNA and RNA as well as at least a qualitative understanding of the structure and stability of mismatches. Absorbance melting experiments are presented on oligonucleotides containing P:Z base pairs and P:C, P:T, Z:G, and Z:A mismatches, providing DDH , DDS , and DDG 37 values for the modified base pairs. P:Z forms a slightly more stable base pair than G:C, primarily due to entropic stabilization, and the G:Z mismatch is significantly more stable than G:T. Wobble geometries are proposed for the G:Z and P:C mismatches. This work demonstrates that substitution of P for G and Z for T can be a useful tool for the control of the relative thermodynamic stabilities of representative designed secondary structures. 395a 1986-Pos Board B123 Binding Studies of Small Molecules to Telomeric Quadruplex DNA for Targeted Singlet Oxygen Production Yasemin Kopkalli1, Craig Biegel1, Ryan Khemraj1, Lesley Davenport2. 1 Chemistry, Brooklyn College of the City University of New York, Brooklyn, NY, USA, 2Chemistry, Brooklyn College and The Graduate Center of the City University of New York, Brooklyn, NY, USA. Recent studies [Shieh et al., ACSNano (2010), 4, 1433; Yin et al., Chem. Comm (2012), 48, 6556] have suggested that G-quadruplexed DNA (qDNA) structures can serve as carriers for targeted delivery of photosensitizers in photodynamic therapies. In our studies, we have investigated the in vitro singlet oxygen production from the photosensitive porphyrins, N-methyl mesoporphyrin IX (NMM) and 5,10,15,20-tetrakis(N-methyl-4-pyridyl)21H,23H-porphine tetratosylate (TMPyP4), when bound to both calf thymus duplex DNA (ct-DNA) and Kþ-stabilized G-quadruplexed human (hTTAGGG)4 telomeric DNA (qDNA). Competition micro-dialysis assays confirmed selective binding of NMM to the mixed hybrid qDNA conformation over TMPyP4, which binds non-selectively to both qDNA and ct-DNA. Sensitized singlet oxygen production was detected using the fluorescent singlet oxygen sensor green (SOSG) assay immediately following porphyrin-DNA irradiation. First-order singlet oxygen generation was detected for both NMM and TMPyP4 when bound to qDNA. However, no appreciable singlet oxygen production was detected for NMM when bound to ct-DNA suggesting potential targeted chemotherapeutic applications for NMM bound to qDNA. CD-studies suggest no change in conformation of the qDNA following irradiation. Currently, we are investigating the effect of the Naþ-stabilized antiparallel qDNA conformation on rate and efficiency of singlet oxygen production from these porphyrins. (Supported by NIHSCORE GM 095437-04.) 1987-Pos Board B124 Regulation of the 3’ UTR in BDNF mRNA at the DNA Level Brett A. DeMarco. Chemistry and Biochemistry, Duquesne University, Pittsburgh, PA, USA. Brain-derived neurotrophic factor (BDNF) is part of the neurotrophic family of genes that encodes for proteins that are known to promote survival of neurons in the peripheral and central nervous systems. The expression of the BDNF gene results in the production of two mRNAs with different lengths, one with a short 3’ untranslated region (UTR) and the second with a long 3’ -UTR, which are believed to serve different functionalities. The ratio of long to short 3’ -UTR in BDNF mRNA changes in different regions of the brain. The long 3’ -UTR mRNA is found in both dendrites and soma, whereas the short 3’ -UTR is only found in the soma, implying that the BDNF mRNA with the long 3’ -UTR is involved in maintaining the neuroplasticity of the brain. Thus, the regulation of the BDNF 3’ -UTR length is important, and it may be achieved at the DNA level, at the mRNA level or both. In this study we are investigating the potential of a 20 nucleotide DNA sequence located in the BDNF gene to adopt non-canonical G quadruplex and i-motif structures that might be involved in regulating the production of long versus short length BDNF mRNA 30 -UTR. 1988-Pos Board B125 Quantitative Investigation of the Role of SeqA in Escherichia Coli Chromosome Segregation Julie A. Cass, Nathan J. Kuwada, Paul A. Wiggins. Physics, University of Washington, Seattle, WA, USA. Although many bacterial species possess a set of par genes responsible for chromosome segregation, Escherichia coli and a number of other gram-negative bacteria appear to be missing the canonical par genes. Nevertheless, these bacteria do possess a unique set of genes that are found only in species that lack the canonical partitioning system, suggesting that a subset of these genes may have superseded their function. Among this subset is the DNA-binding protein SeqA, already known to play a central role in the regulation of DNA replication. Combining high-throughput fluorescence microscopy and automated image analysis, we are investigating the role of SeqA in segregation by two complementary approaches: (1) characterizing correlated dynamics of SeqA and the origin of replication by simultaneous visualization of SeqA and oriC localization throughout the segregation process in hundreds of independent cells, and (2) quantitative analysis of chromosome dynamics in a SeqA mutant to estimate the magnitude of the biasing forces during the segregation process with and without SeqA. We believe this quantitative imaging-based approach will provide new insights into the segregation process in E. coli which has so far thwarted traditional genetic approaches. 396a Tuesday, February 10, 2014 1989-Pos Board B126 Packing and Phase Transitions in DNA Duplexes and Tetraplexes: Similarities and Differences Selcuk Yasar, Rudolf Podgornik, V. Adrian Parsegian. University of Massachusetts, Amherst, MA, USA. We have mapped the thermodynamic potentials that drive transitions of DNA in uni- and di-valent salt solutions. The successive mesophases, with measured free energies of deformation and transition, allow computation of interaction potentials as well as transition entropies and enthalpies. We have been able to measure transitions of DNA tetraplexes and duplexes and to compare entropic and enthalpic contributions. Changes in fluctuation free energies are much greater at DNA-ordering transitions for tetraplexes than for duplexes, indicating strong entropic contributions. Disordering due to fluctuations is much greater in the less-ordered (cholesteric) phase, seen in broadening of x-ray scattering peaks. This indicates attraction in the more-ordered phase, where packing is stricter, and the effect of fluctuations is much smaller. This attraction is stronger for quadruplexes than for duplexes. We can read two kinds of information from the x-ray data: the degree of ordering, and the change in density. These changes are much bigger in tetraplexes than in double helical DNA. In addition, we also observe that upon decreasing the applied osmotic stress on the less-ordered phase, there is spontaneous disassembly of tetraplexes. This second transition, from a stack of tetramers to monomers, also depends on temperature, allowing us again to measure transition entropy and free energy. Lowering the temperature in the cholesteric phase favors tetraplex formation. The critical osmotic pressure for the formation of tetraplexes, just as the critical osmotic pressure for inducing the higher-density packing, depends strongly on the temperature. Out next goal is to compare tetraplexes, which lack a linking backbone, with quadruplexes (where the bases are linked) so as to see the stabilizing contributions of a polymer backbone. 1990-Pos Board B127 The Relationship between Electrophoretic Mobility and Polyelectrolyte Charge Nancy C. Stellwagen. Department of Biochemistry, University of Iowa, Iowa City, IA, USA. Many textbooks state that the electrophoretic mobility of a polyelectrolyte is proportional to its effective charge divided by its frictional coefficient. We have tested this relationship by analyzing the mobilities of single- and double-stranded DNA molecules containing the same number of bases or base pairs and different numbers of negatively charged phosphate linkers. Small organic molecules containing different numbers of charged residues were also analyzed,using data taken from the literature. In each case, the free solution mobilities of the charge variants of a given molecule were divided by the mobility of the charge variant with the highest number of charged residues, measured under the same conditions. The mobility ratios were then plotted as a function of the fractional charge of each molecule. The results indicate that the fractional mobilities of polyelectrolytes of the same size are proportional to the logarithm of the fractional charge, not the first power of the charge as commonly assumed. The Manning theory of DNA electrophoresis and electrophoretic theories based on the zeta potential both predict a logarithmic dependence of the mobility on charge density. The experimental mobility ratios will be compared with the predictions of these two theories. 1991-Pos Board B128 Enhanced Sampling of DNA Step Parameters: Impact of Methylation on DNA Shape and Flexibility Aleksandra Karolak, Arjan van der Vaart. Chemistry, USF, Tampa, FL, USA. We present a novel approach for the selection of DNA step parameters as reaction coordinates in umbrella sampling simulations. Simplified representation of DNA that uses only three atoms per base, allowed for highly efficient calculations of the step parameters and their Cartesian derivatives in the molecular dynamics simulations. Good correlation between the actual and calculated twist, roll, tilt, rise, slide and shift was obtained. The method is illustrated through its application to the unmethylated and methylated DNA systems. Impact of the methylation on the shape and flexibility of DNA depending on the location of the methyl group is discussed. 1992-Pos Board B129 Three-Dimensional Modeling of Single Stranded Hairpin DNA Aptamers Iman Jeddi, Leonor Saiz. Biomedical Engineering, University of California, Davis, Davis, CA, USA. Aptamers are short oligonucleotides that are selected for affinity binding to a wide range of targets and provide a number of advantages over antibodies including robustness, low cost, and reusability [1,2]. The relatively simple chemical structure of aptamers allows the insertion of electrochemical or fluorescent reporter molecules as well as surface-binding agents in specific locations on the oligonucleotide. During probe-target binding, the conformation change of the aptamer may be exploited to generate an analytical signal. The robustness and simplicity of aptamers has allowed for multiple uses of aptamer-based biosensors and a number of direct detection strategies employing aptamers have been proposed. However, in order for commercialization of these devices to become feasible, significant improvements in optimization for consistency and reproducibility must be done. Overcoming these challenges has been hampered by a lack of complete understanding of the molecularlevel biophysics involved [3]. In this regard, computational studies can complement experimental studies in improving our understanding about the structure, molecular-level interactions, dynamics, and solvent effects of biomolecular complexes [4,5]. Here, we present a method to predict the three-dimensional structure of single stranded DNA aptamers from their sequence. References: [1] Ellington AD & Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346: 818-822. [2] Liu Y et al. (2010) Aptamer-based electrochemical biosensor for interferon gamma detection. Analytical chemistry 82: 8131-8136. [3] Zhou W et al. (2014) Aptamer-based biosensors for biomedical diagnostics. The Analyst 139: 2627-2640. [4] Saiz L (2012) The physics of protein-DNA interaction networks in the control of gene expression. Journal of Physics: Condensed Matter 24: 193102. [5] Sinha SK & Saiz L (2014) Determinants of protein-ligand complex formation in the thyroid hormone receptor a: a Molecular Dynamics simulation study. Computational and Theoretical Chemistry 1038, 57-66. 1993-Pos Board B130 Resolving the DNA Binding Mode of a Rotationally Flexible Binuclear Ruthenium Complex Ali A. Almaqwashi1, Johanna Andersson2,3, Per Lincoln3, Ioulia Rouzina4, Fredrik Westerlund3, Mark C. Williams1. 1 Department of Physics, Northeastern University, Boston, MA, USA, 2 Department of Chemistry-BMC, Uppsala University, Uppsala, Sweden, 3 Department of Chemical and Biological Engineering, Chalmers University of Technology, Gothenburg, Sweden, 4Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN, USA. Binuclear ruthenium complexes demonstrate slow dissociation upon binding to DNA, and they are considered as potential DNA-targeted therapeutic drugs. Quantitative investigations of their properties, such as binding affinity, binding mode and kinetics, require approaches beyond traditional experimental techniques. In particular, previous reports showed that the rotationally flexible binuclear ruthenium complex D,D-[m-bipb(phen)4Ru2]4þ (D,D-Pi) strongly condenses DNA. However, the strong DNA condensation poses a challenge in bulk experiments such that even the DNA-ligand binding mode could not be resolved. We examined the kinetics of DD-Pi interactions with single lDNA molecule as a function of a constant applied force of 30 pN and ligand concentration of 5 nM using dual-beams optical tweezers. We find that D,DPi exhibits characteristics of threading intercalation into DNA base pairs. DNA elongation measurements, as monitored over tens of minutes, illustrate two distinct phases during association; rapid intercalation that is analogous to classic intercalation, followed by very slow intercalation that approaches equilibrium with a rate that is comparable to that observed for other threading intercalators. We investigated DD-Pi dissociation after reaching the equilibrium extension by rinsing the binding ligands from the surrounding solution, and observed that the DNA-ligand complex extension decreases to the DNAonly extension over a timescale longer than the association process. Interestingly, the dissociation measurements fit well to a single rate that is slower than the dissociation rate measured for the previously reported binuclear ruthenium complex D,D-[m-bidppz-(phen)4Ru2]4þ (D,D-P), which is a threading intercalator. Further measurements of force-dependent intercalation thermodynamics and kinetics will allow us to fully quantitatively characterize the complex binding mechanism of this unusual intercalator. Tuesday, February 10, 2014 1994-Pos Board B131 Epigenetics and Other Factors that Affect Folding and Stability of DNA I-Motif Structures Samantha M. Reilly1, Yogini P. Bhavsar-Jog1, Sara E. Wingate1, Daniel F. Lyons2, Robert T. Wright2, Tracy A. Brooks3, John J. Correia2, David M. Jameson4, Randy M. Wadkins1. 1 Dept. of Chemistry and Biochemistry, University of Mississippi, University, MS, USA, 2Dept. of Biochemistry, University of Mississippi Medical Center, Jackson, MS, USA, 3Dept. of BioMolecular Sciences, University of Mississippi, University, MS, USA, 4Dept. of Cell and Molecular Biology, University of Hawaii at Manoa, Honolulu, HI, USA. The four-stranded i-motif (iM) conformation of cytosine-rich DNA has importance to a wide variety of biochemical systems that range from their use in nanomaterials to potential roles in oncogene regulation. The iM structure is formed at slightly acidic pH, where hemi-protonation of cytosine results in a stable C-Cþ base pair. Fundamental studies to understand iM formation from C-rich strands of DNA are described. We present a systematic characterization of the consequences of epigenetic modifications, molecular crowding, degree of hydration, and DNA sequence on the stabilities of iM-forming sequences. We used a number of biophysical techniques to characterize both the folded iM and the folding kinetics of an iM. We established a mechanism for the folding. We observed that the C-Cþ hydrogen bonding of certain bases initiates the folding of the iM structure. We also observed that substitutions in the loop regions of iMs give a distinctly different kinetic signature during folding as compared to those bases that are intercalated. Our data reveal that the iM passes through a distinct intermediate form between the unfolded and folded form. In the course of determining this folding pathway, we established that the fluorescent dC analogs tC and PdC can be used to monitor individual residues of an iM structure and can be used to determine the pKa of an iM. Our results indicate that 5-hydroxymethylation of cytosine destabilized the iMs against thermal and pH-dependent melting, while 5-methylcytosine modification stabilized the iMs. Under molecular crowding conditions, the thermal stability of iMs increased and the pKa was raised to near 7.0. Taken together, our work has laid the foundation for examining folding and structural changes in more complex iMs. Protein-Nucleic Acids II 1995-Pos Board B132 Activation of PKR by Stem-Loop Rnas with Flanking ssRNA Tails Christopher Mayo, Prisma Lopez, James Cole. University of Connecticut, Storrs, CT, USA. Protein Kinase R (PKR) is a central component of the innate immunity antiviral pathway and is activated by double stranded RNA (dsRNA). PKR contains a Cterminal kinase domain and two tandem dsRNA binding motifs. In the accepted model for activation, binding of multiple PKR monomers to dsRNA enhances dimerization of the kinase domain. A minimum dsRNA length of 30 bp is required for binding two PKR monomers and eliciting strong enzymatic activation. However, short (15 bp) stem-loop RNAs containing flanking single stranded tails (ss-dsRNAs) are capable of activating PKR. Activation requires a 50 -triphosphate, the presence of both 5’ and 3’ ssRNA tails, and a tetraloop capping the duplex stem yet how these moieties modulate protein binding and enzymatic activity remains unknown. We have incorporated a photoactivatable unnatural amino acid into PKR’s dsRNA binding domain and used a novel crosslinking protocol to gain insight into how these structural features orient binding of PKR to the ss-dsRNA. 1996-Pos Board B133 Determining the DNA Diffusion Behavior of SA2 on Various DNA Substrates Preston J. Countryman1, Jiangguo Lin1, Parminder Kaur1, Edward Brennan1, Haijiang Chen2, Changjiang You3, Jacob Piehler3, Yizhi Jane Tao2, Hong Wang1. 1 Physics, North Carolina State University, Raleigh, NC, USA, 2Biochemistry and Cell Biology, Rice University, Houston, TX, USA, 3Biophysics, Universita¨t Osnabru¨ck, Osnabru¨ck, Germany. Cohesin is a multi-protein complex involved in sister chromatid cohesion during cell replication and double strand DNA break repair. Cohesin core complex consists of a ring-like trimer and either SA1 or SA2 in somatic vertebrate cells. While SA1 and SA2 share ~70% homology, only SA1 contains a critical AT hook domain responsible for its binding to telomere sequences. Cohesin-SA1 holds sister chromatids together at telomere regions during cell separation and can be found at specific promoter regions, while Cohesin-SA2 is predominantly located at intergenic and centromere regions. The mechanism by which Cohesin locates specific DNA sequences is currently unknown. To understand 397a the role that SA1 or SA2 has in Cohesin/DNA interactions, we used the singlemolecule techniques atomic force microscopy (AFM) and fluorescence imaging of quantum dot labeled proteins on DNA tightropes. Preliminary data indicates that SA1 carries out 1-D diffusion on DNA, binds with high affinity to telomeric sequences, and pauses on telomeric and promoter regions. In contrast, SA2 exhibits static and dynamic populations without pausing for telomere, centromere, and promoter DNA sequences. We propose that 1-D sliding and sequence dependent pausing by SA1 provides binding specificity and stability during the cohesion process at telomeres, while.SA2 alone, lacking the AT hook domain, uses different DNA binding mechanisms. 1997-Pos Board B134 A Human Transcription Factor in Search Mode Kevin Hauser1, Bernard Essuman2, Evangelos Coutsias3, Miguel GarciaDiaz4, Carlos Simmerling1. 1 Chemistry, Stony Brook University, Stony Brook, NY, USA, 2Chemistry, Suffolk County Community College, Selden, NY, USA, 3Applied Mathematics & Statistics, Stony Brook University, Stony Brook, NY, USA, 4 Department of Pharmacological Sciences, Stony Brook University, Stony Brook, NY, USA. Transcription factors (TF) change shape to search and recognize DNA, shifting the energy landscape from a weak binding mode to a tight binding mode. But the mechanism of TF conformational change, which deforms DNA during recognition, remains unresolved. Superhelical TFs have a modular helical topology that track along the DNA helical groove. Thus, TF and DNA helical pitch match. Our goal is to develop a mechanism of TF-DNA search and recognition using a superhelical TF as a model. Human MTERF1 is a superhelical TF that terminates transcription by unwinding mitochondrial DNA. The structure of apoMTERF1 is likely flexible: few packing interactions between modules and we are unable to crystallize it. We hypothesize that apoMTERF1 can have low strain conformations with low helical pitch that can track a B-DNA major groove. To characterize apoMTERF1, we used a coarse grained simulation to model intrinsic motions and atomistic MD to obtain a diverse structural ensemble. The coarse grained simulation showed the largest mode of apoMTERF1 is a pitching motion. The MD showed apoMTERF1 is flexible, and unstrained in a conformation with low pitch that matches B-DNA. To show that low pitch apoMTERF1 structures were stable in search mode, we docked these structures to B-DNA and equilibrated the complexes using atomistic MD. The search mode complexes were stable: decreasing in energy and increasing in structural complementarity. Surprisingly, the helical motion from the coarse grained and atomistic simulations was also present in the search mode. The helical motion permits the individual motifs to shift along the sequence in a stepping translocation process and might also be involved in unwinding DNA during recognition. We are currently modeling the transition to recognition mode to test if the mechanism can be explained by MTERF1 natural helical motions. 1998-Pos Board B135 Two-Step DNA Intercalation by Threading of the Flexible Ruthenium Dimer Studied by the Single Molecule DNA Stretching Ioulia Rouzina1, Meriem Bahira2, Ali Almaqwashi2, Micah McCauley2, Fredrik Westerlund3, Mark C. Williams2. 1 University of Minnesota, Minneapolis, MN, USA, 2Department of Physics, Northeastern University, Boston, MA, USA, 3Department of Chemical and Biological Engineering, Chalmers University of Technology, Gothenberg, Sweden. Ruthenium complexes are small synthetic molecules with a wide range of possible uses, including cancer therapy and sensitive fluorescent markers for duplex DNA binding. The ruthenium complex [m-C4(cpdppz)2(phen)4Ru2] 4þ has been engineered to have a high affinity for DNA and a low dissociation rate. The complex consists of two Ru(phen)2dppz2þ moieties connected by a flexible linker. However, the mechanism by which these molecules interact with DNA is not well understood. To quantify these interactions, doublestranded DNA is stretched with optical tweezers, and exposed to the ligand under a fixed applied force. When binding to DNA, the two Ru(phen)2dppz 2þ moieties intercalate between base pairs via a threading. We find that the ligand association can only be described by a two-exponential process, indicating multi-step binding. By measuring the concentration dependence of the fast and slow binding modes at several forces and fitting this dependence to a three state kinetic model, we show that the fast mode is bimolecular intercalation of the first dppz moiety, in pre-equilibrium to the ~10-fold slower intercalation of the second dppz moiety of the same flex-Ru2 molecule. We characterize forcedependence of each rate and DNA elongations associated with each transition state. We estimate the zero-force binding kinetics and equilibrium binding constants for each of the two intercalations steps and of the complete binding 398a Tuesday, February 10, 2014 process by extrapolating our measured force dependence of these parameters to the force-free state. We conclude that at zero force the [m-C4(cpdppz)2(phen) 4Ru2]4þ binding involves fast (~20 s) association, slow (~600 s) dissociation, and very tight (Kd~10 nM) binding. The methodology developed in this work will be useful for studying other slowly intercalating ligands and proteins. 1999-Pos Board B136 The Role of the Threading Moiety in DNA Threading Intercalation by Ruthenium Dimer Complexes Andrew G. Clark1, Thayaparan Paramanathan1,2, Fredrik Westerlund3, Per Lincoln3, Micah J. McCauley1, Ioulia Rouzina4, Mark C. Williams1. 1 Physics, Northeastern University, Boston, MA, USA, 2Physics, Bridgewater State University, Bridgewater, MA, USA, 3Chemical and Biological Engineering, Chalmers University of Technology, Gothenburg, Sweden, 4 Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, USA. There is significant interest in ruthenium dimer complexes due to their novel DNA binding properties. The binuclear ruthenium complex DD-[mbidppz(bipy)4Ru2]4þ that we investigate in this study binds to DNA by threading intercalation, in which one of the bulky ruthenium moieties threads through the DNA base pair stack to reach its bound state. The extremely slow dissociation from DNA of this threading intercalator gives it properties needed for chemotherapy drugs, while also making it difficult to study in traditional bulk experiments. In this study we use optical tweezers in order to quantify the threading kinetics as well as the equilibrium behavior of this ruthenium dimer complex to understand how changing the ancillary ligands coordinated to ruthenium (threading moiety) affects the process of threading. To observe the slow threading process, the DNA was held at a constant force in the presence of the ligand and the DNA extension was measured as a function of time until the extension reached equilibrium. When the concentration of the ligand is increased during these constant force measurements, the total extension of the DNA will also increase until it becomes saturated at some concentration as all of the ligand binding sites are filled. These measurements of the equilibrium DNA extension allow for a full quantitative description of the binding kinetics and equilibrium behavior of the complex. The preliminary data suggests that the DD-[m-bidppz(bipy)4Ru2]4þ complex with its bicyclic bipyridine ligand, shows significantly faster kinetics compared to the DD-[m-bidppz(phen)4 Ru2]4þ , complex with a tricyclic phenanthroline ligand counterpart. 2000-Pos Board B137 Molecular Interaction between Escherichia Coli Topoisomerase I and pBAD/Thio Supercoiled Plasmid DNA Purushottam Tiwari1, Thirunavukkarasu Annamalai2, Bokun Cheng3, Gagandeep Narula2, Xuewen Wang1, Yuk-Ching Tse-Dinh2, Jin He1, Yesim Darici1. 1 Department of Physics, Florida International University, Miami, FL, USA, 2 Department of Chemistry and Biochemistry, Florida International University, Miami, FL, USA, 3Department of Biochemistry and Molecular Biology, New York Medical College, Valhalla, NY, USA. DNA topoisomerase regulates the topological state of DNA, which is very important for replication, transcription and recombination. The relaxation of negatively supercoiled DNA is catalyzed by bacterial DNA topoisomerase I (topoI) and this reaction requires Mg2þ. We quantitatively studied the molecular interactions between Escherichia coli topoisomerase I (EctopoI) and pBAD/Thio supercoiled plasmid DNA using surface plasmon resonance (SPR) technique. The equilibrium dissociation constant (Kd) for EctopoIpBAD/Thio interactions is determined to be about 8 nM. A slightly higher Kd value (~15 nM) was obtained for Mg2þ coordinated EctopoI (Mg2þEctopoI)-pBAD/Thio interactions. In addition, we observed a larger dissociation rate constant (kd) for Mg2þEctopoI-pBAD/Thio interactions (~0.043 s1), compared to EctopoI-pBAD/Thio interactions (~0.017 s1). These results suggest that enzyme turnover during plasmid DNA relaxation is enhanced due to the presence of Mg2þ. 2001-Pos Board B138 Force Regulated Association Dynamics of RPA on Forked DNA Felix E. Kemmerich1, Peter Daldrop1, Maryna Levikova2, Petr Cejka2, Ralf Seidel1. 1 Single molecule analysis group, University of Mu¨nster, Mu¨nster, Germany, 2 Institute of Molecular Cancer Research, University of Zurich, Zurich, Switzerland. Replication protein A (RPA) is involved in virtually all aspects of eukaryotic DNA processing, including replication, recombination and repair. The strong binding of RPA to single-stranded DNA (ssDNA), which provides protection from nucleolytic cleavage, is well established. Binding to partially doublestranded DNA (dsDNA) and disruption of DNA intermediates possessing sec- ondary structures was reported previously, however the underlying mechanism remains unclear. Utilizing single-molecule magnetic tweezers, we now show that both human and yeast RPA can open a DNA hairpin subjected to force. For this welldefined substrate geometry which closely mimics a replication fork, we measured the force-dependent RPA association and dissociation, whilst resolving individual binding events. To explain the observed helix opening activity, we propose a passive model: Transient openings of the DNA helix, enhanced by stronger forces, become trapped by RPA binding. When the force is reduced, dissociation is driven by helix rehybridization. This leads to an exponential dependence of the association and dissociation rates on the applied force, in agreement with our data. By extrapolating our measured rates, we conjecture that RPA does not disrupt dsDNA on its own in the absence of force, but rather seems to rely on the active unwinding of a helicase. RPA could then strongly bind and protect ssDNA produced in its wake, providing a scaffold for the recruitment of further DNA processing enzymes. While the fork is held open, changes to the force exerted by downstream enzymes may regulate the degree of RPA binding on the ssDNA. Upon completion of the DNA processing step the fork would become unblocked. Rapid helix rehybridization then expels RPA from the ssDNA with dissociation rates on the order of hundreds of base-pairs per second, such that the completely processed dsDNA is returned to its native RPA-unbound state. 2002-Pos Board B139 Specific Binding of the Nucleocapsid Protein Transforms the Folding Landscape of the HIV-1 TAR RNA Hairpin Micah J. McCauley1, Ioulia Rouzina2, Kelly Hoadley2, Robert J. Gorelick3, Karin Musier-Forsyth4, Mark C. Williams1. 1 Physics, Northeastern University, Boston, MA, USA, 2Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, USA, 3AIDS and Cancer Virus Program, SAIC-Frederick, Inc., Frederick, MD, USA, 4Chemistry and Biochemistry, The Ohio State University, Columbus, OH, USA. Retroviral nucleocapsid (NC) proteins are nucleic acid chaperones that play a key role in the viral life cycle. During reverse transcription, HIV-1 NC destabilizes nucleic acids, including the transactivation response (TAR) hairpin to facilitate their structural re-arrangement into the lowest free energy conformations. By combining single molecule optical tweezers measurements with mfold-based free energy calculations of the unfolding landscape, we determine the equilibrium TAR stability and characterize the unfolding transition state in both the presence and absence of NC. Our results show that protein binding occurs specifically at guanines that border structural defects in the upper part of TAR hairpin. This results in preferential destabilization by NC of this part of TAR, thereby shifting the transition state closer to the bottom of the TAR stem, leading to a shorter critical unfolding length. Thus, NC facilitates TAR RNA annealing to its complementary DNA hairpin while having little effect on the stability of the final annealed duplexes. These results provide the first direct measurement of alterations in an RNA folding landscape that are induced by protein-RNA interactions, and are a novel case study of the effect of a protein that specifically destabilizes particular elements of the nucleic acid secondary structure. 2003-Pos Board B140 Factors that Influence PKR Dimerization and Activation Bushra Husain, Michael Bruno, Matthew Angeliadis, James Cole. Molecular and Cell Biology, University of Connecticut, Storrs, CT, USA. Protein kinase R (PKR) is a key component of the interferon-induced innate immunity pathway. It is expressed in a latent, inactive form and is activated upon binding dsRNA and subsequent autophosphorylation. PKR contains a C-terminal kinase domain and two tandem dsRNA binding motifs (dsRBMs) at the N-terminus. These regions are separated by a 90-residue, unstructured linker. Dimerization is believed to play a critical role in the mechanism of PKR activation by dsRNA. Activation requires dsRNAs long enough to accommodate two PKR monomers. However, there is no direct evidence for dimerization of PKR on dsRNA lattices. Under certain conditions, shorter RNAs bind multiple PKRs but fail to activate, suggesting that additional factors are involved. It is believed that binding of the second dsRBM is required for PKR activation. We have characterized the role of the second dsRBM by introducing mutations that block its interaction with dsRNA. The distance dependence of PKR dimerization and activation were probed using synthetic RNAs that function as molecular rulers. These molecules consist of two 15 bp duplex regions separated by variable length regions of 20 -O-methyl modified dsRNA that act as rigid and inert barriers. The effect of linker length was investigated using a PKR homolog containing a short, 25-residue linker. We developed a fluorescence assay to Tuesday, February 10, 2014 quantitatively probe PKR dimerization upon binding to RNA. The fluorescence anisotropy of a probe placed near the kinase dimer interface decreases upon PKR binding to an activating 40 bp dsRNA due to depolarization induced by homo-FRET. Thus, the kinase domains form a dimer when two PKR monomers bind to this dsRNA. Surprisingly, several non-activating dsRNAs also induce dimerization, suggesting that PKR dimerization is necessary but not sufficient for activation. 2004-Pos Board B141 Quantitative DNA Binding, Looping, and Compaction Properties of the HIV-1 Viral Protein R Divakaran Murugesapillai1, Micah J. McCauley1, Ioulia Rouzina2, Serge Bouaziz3, Mark C. Williams1. 1 Department of Physics, Northeastern University, Boston, MA, USA, 2 Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, USA, 3Faculte´ de Pharmacie, Universite´ Paris Descartes, Paris, France. Human immunodeficiency virus type 1 (HIV-1) Viral protein R (Vpr) is packaged into virions (~200 molecules) and is essential for viral replication. While several in vivo functions have been attributed to Vpr, one primary function is believed to be transport of the HIV-1 pre-integration complex into the nucleus. Because the nuclear pore diameter is approximately 25 nm, the DNA must be highly compact in order to enter the nucleus. To understand the mechanism by which Vpr may facilitate this nuclear transport, we combine single molecule stretching and atomic force microscopy (AFM). We measure the DNA binding affinity of Vpr for the first time. We then investigate the ability of Vpr to both bind and compact DNA. To do this, we hold DNA at low force for fixed times, allowing it to form loops and other compact structures. We find that the timescale required for the formation of large loops due to protein-DNA bridging is several minutes. In contrast, by holding the DNA molecule at a constant force of 15 pN and measuring its length change, Vpr can also actively compact DNA on the timescale of tens of seconds (15 5 2 s), representing a different compaction process involving DNA shortening likely due to Vpr binding alone. The persistence length determined from AFM at a low concentration is much longer than that of bare DNA, suggesting that Vpr forms shorter but more rigid structures upon binding DNA. AFM images also demonstrate DNA bridging between strands, consistent with the looping observed in optical tweezers experiments. These results support a model in which Vpr translocates the viral DNA into the nucleus by forming compact structures mediated by protein-DNA and protein-protein interactions. 2005-Pos Board B142 Repetitive Single-Molecule FRET Fluctuations upon T4 Gene 32 Protein Binding to Single-Stranded DNA Wonbae Lee1,2, John P. Gillies3, Davis Jose2, Peter H. von Hippel2, Andrew H. Marcus1,2. 1 Oregon Center for Optics, Univeristy of Oregon, Eugene, OR, USA, 2 Institute of Molecular Biology and Department of Chemistry, Eugene, OR, USA, 3Department of Chemistry and Biochemistry, Univeristy of Oregon, Eugene, OR, USA. Bacteriophage T4-coded gene 32 protein (gp32) is an essential component of the T4 DNA replication, recombination, and repair systems. Gp32 is a single-stranded DNA binding protein that binds cooperatively to singlestranded (ss) DNA templates exposed by the function of the T4 primosome helicase, and configures these templates for efficient use by the replisome. To this end gp32 binding helps melt out transiently formed ssDNA secondary structures, protects exposed ssDNA sequences from nucleases, and helps to integrate the functions of the other components of the replication complex. It has been well studied by bulk biochemical methods for about 40 years. However, many aspects of the detailed mechanisms – and especially the dynamics – of the interactions of gp32 with both double-stranded (ds) and single-stranded DNA are still not well studied or well understood. We have used single-molecule Fo¨rster Resonance Energy Transfer (sm-FRET) methods to monitor the cooperative and non-cooperative binding of gp32 to the ssDNA sequences of model DNA replication forks, and have shown that the observed binding dynamics depend significantly on gp32 concentration, salt concentration and ssDNA segment length. Upon addition of 1 mM concentrations of gp32 into a sample chamber containing tethered single molecule replication fork constructs, we observed the appearance of ‘repetitive FRET fluctuations’ of the individual DNA molecules (>70% of the molecules per imaging area) on a 100 ms timescale. We noted also that these repetitive FRET fluctuations were substantially less prominent under the conditions of tight and fully cooperative gp32 protein binding. We have used ensemble and single-molecule fluorescence approaches to probe the dynamics of these gp32-ssDNA interactions in detail, and will discuss possible molecular inter- 399a pretations of these smFRET fluctuations in terms of potential reaction intermediates and association-dissociation pathways in real time. 2006-Pos Board B143 How MeCP2 and R.DpnI Proteins Recognize Methylated DNA Volkhard Helms, Siba Shanak. Center for Bioinformatics, Saarland University, Saarbru¨cken, Germany. DNA methylation plays a major role in organismal development and the regulation of gene expression. Methylation of cytosine bases and its cellular roles in eukaryotes are well established, as well as methylation of adenine bases in bacterial genomes. Here, we present results from molecular dynamics simulations, alchemical free energy perturbation, and MM-PBSA calculations to explain the specificity of the R.DpnI enzyme for binding to adenine-methylated DNA in both its catalytic and winged-helix domains. We find that adeninemethylated DNA binds more favorably to the catalytic subunit of R.DpnI (4 kcal/mol) and to the winged-helix domain (1.6 kcal/mol) than unmethylated DNA. In particular, N6-adenine methylation is found to enthalpically stabilize binding to R.DpnI. In contrast, C5-cytosine methylation stabilizes binding to the MBD domain of the MeCP2 entropically with almost no difference in binding enthalpy. 2007-Pos Board B144 DNA Looping and Genome Architecture: How Proteins can Connect and Organize Chromosomes Nicolas Clauvelin, Wilma K. Olson. BioMaPS Institute, Rutgers University, Piscataway, NJ, USA. The control of gene expression sometimes entails the folding of DNA into looped structures mediated by the binding of protein. Although the literature abounds with examples of single DNA loops induced by the attachment of sequentially distant genetic elements on a common protein core, recent studies have demonstrated the occurrence of multiple loops formed by three or more remote, protein-anchored sites. For example, the Escherichia coli Gal repressor has the ability to form oligomeric structures leading to higher-order helical protein pathways that can secure multiple chromosomal connections. Moreover, several novel experimental investigations have highlighted the role of bridging proteins, such as the macrodomain Ter protein (MatP) and the histone-like structuring protein (H-NS), in chromosome condensation and organization. These proteins are thought to be able to bridge distant DNA sites and to participate in the folding of the bacterial genome. We are examining the entanglement of DNA loops that attach to such proteins with the help of a novel energy minimization method associated with traditional Monte Carlo approaches. We focus on the multiple loops that can be induced by oligomeric Gal assemblies and report the relevant energy landscapes and topological and statistical properties as functions of the number of Gal repressors and the chain lengths of the different loops. In addition, we take advantage of the fact that our optimization method accounts for the presence along DNA of bound ligands to reveal how the binding of architectural proteins (e.g., the Escherichia coli histone-like HU protein) can ease or suppress the formation of such loops. Finally, we examine the influence of MatP and H-NS on the conformation and fluctuations of DNA minicircles to understand how these proteins may contribute towards the formation of topological domains. 2008-Pos Board B145 Common Aspects of G-Quadruplex Destabilization among Helicases and Single Stranded DNA Binding Proteins Jagat B. Budhathoki1, Sujay Ray1, Pavel Janscak2, Jaya Yodh3, Hamza Balci1. 1 Dept of Physics, Kent State University, Kent, OH, USA, 2Institute of Molecular Cancer Research, University of Zurich, Zurich, Switzerland, 3Dept of Physics, University Of Illinois, Urbana-Champaign, IL, USA. Various helicases and single-stranded DNA (ssDNA) binding proteins are known to destabilize G-quadruplex (GQ) structures, which otherwise result in genomic instability. Bulk biochemical studies have shown that Bloom helicase (BLM) unfolds both intermolecular and intramolecular GQ in the presence of ATP. Using single molecule FRET, we show that binding of BLM to ssDNA in the vicinity of an intramolecular GQ leads to unfolding of the GQ in the absence of ATP. We show that the efficiency of BLM-mediated GQ unfolding correlates with the binding stability of BLM to ssDNA overhang, as modulated by the nucleotide state, ionic conditions, overhang length, and overhang directionality. A related protein WRN showed similar activity to BLM. These results are surprisingly similar to those we observed on interactions of ssDNA binding protein Replication Protein A with GQ, which also does not require ATP or enzymatic activity to unfold GQ. These similarities point out to common features of GQ destabilization mechanisms of helicases and ssDNA binding 400a Tuesday, February 10, 2014 proteins, in which binding of the protein is what initiates and in some instances is adequate to unfold the GQ. 2009-Pos Board B146 Dynamic Interactions between DNA and the T4 Single-Stranded Binding Protein gp32: Multi-Dimensional Correlation Analysis of Microsecond Single-Molecule FRET and Linear Dichroism Fluctuations Carey Phelps1, Brett Israels1, Wonbae Lee1, Davis Jose2, Peter H. von Hippel2, Andrew H. Marcus1. 1 Chemistry, University of Oregon, Eugene, OR, USA, 2Molecular Biology, University of Oregon, Eugene, OR, USA. Protein-nucleic acid interactions are of central importance in genomeregulatory processes. The DNA replication system of the T4 bacteriophage is an excellent model system to study DNA replication in higher organisms, since the T4-coded replication machinery utilizes many of the same components. Here we present studies of the kinetics of binding of gp32, the singlestranded (ss) DNA binding protein of the T4 system, whose roles include the functional integration of the other components of the T4 replication complex as well as protecting the transiently-exposed single-stranded DNA template sequences from DNA nucleases and melting out unfavorable secondary structures potentially formed by the lagging strand during DNA replication. We have performed single-molecule measurements of internally-labelled Cy3/Cy5 labeled primer-template DNA constructs in the presence of gp32 by simultaneously monitoring single-molecule Fo¨rster resonance energy transfer (smFRET) and single-molecule fluorescence-detected linear-dichroism (smFLD) on the microsecond time scale. smFRET measurements probe the distance between the fluorophores, while smFLD measurements are sensitive to local orientations of the Cy3-labeled sugar-phosphate backbones of the DNA construct. Multiple transient FRET states are observed when adding gp32 protein to tethered model replication fork DNA constructs, permitting us to track the dynamics of millisecond interconversions between various configurations of the gp32 / ssDNA system. The distribution of FRET states changes as a function of gp32 concentration, suggesting that we may be monitoring fluctuations that reflect changes associated with the progression from isolated to cooperative gp32 binding. We apply a multi-dimensional correlation function analysis to our microsecondresolved smFRET and smLD data to reveal statistically relevant mechanistic information about the reaction pathways of the ssDNA-gp32 system at the replication fork. 2010-Pos Board B147 Exploration of Cytosine Methylation Effects on Protein-DNA Binding Skyler Uhl1, Amber M. Velasco2, Allison M. Nice2, Winston Timp2. 1 Biology, Johns Hopkins University, Baltimore, MD, USA, 2Biomedical Engineering, Johns Hopkins University, Baltimore, MD, USA. Bisulfite sequencing has been employed to great effect to identify DNA methylation changes between different tissues, through stem cell differentiation, and even in cancer development. However, there is an ever increasing problem of assessing the functional importance of these methylation changes- though work has demonstrated that methylation affects protein-DNA interaction, a clear and comprehensive delineation of the strength of these effects is lacking. We employed a modified chromatin immunoprecipitation method combined with bisulfite sequencing to identify the distribution of methylation patterns present in bound DNA. We used both synthetically generated in vitro samples for an unbiased measurement and samples derived from cell lines for physiologically relevant patterns. Our library preparation methodology uses magnetic bead ChIP (Life Technologies) combined with methylated hairpin adapters (NEB) and low-input bisulfite methods (Zymo Research) with custom modifications. We then sequenced these libraries on an Illumina MiSeq for the longread length offered (2x300). The results were compared to the input methylome to determine the relative frequency of methylation patterns bound by protein. In our initial work, we examined the methylation patterns of DNA bound by CTCF and MeCP2. The number of methylated locations required to affect protein-binding was measured, as well as the frequency of binding versus a known binding sequence. We specifically examined DNA upstream and downstream of the known binding motifs, to determine non-local effects of methylation on protein-DNA affinity. 2011-Pos Board B148 Molecular Mechanism of Processive 3’ to 5’ RNA Translocation in the RNA Exosome Complex Lela Vukovic1,2, Debora L. Makino3, Christophe Chipot4,5, Elena Conti3, Klaus Schulten1,5. 1 Department of Physics, University of Illinois at Urbana-Champaign, Urbana, IL, USA, 2Center for the Physics of Living Cells, University of Illinois at Urbana-Champaign, Urbana, IL, USA, 3Department of Structural Cell Biology, Max Planck Institute of Biochemistry, Martinsried, Germany, 4 Laboratoire International Associe´ CNRS-University of Illinois, Universite´ de Lorraine, Vandœuvre-le`s-Nancy, France, 5Beckman Institute, University of Illinois at Urbana-Champaign, Urbana, IL, USA. 3’ to 5’ degradation of a wide range of RNA molecules is performed by the exosome complex, as a key part of cellular quality control. Recent structural studies of this complex revealed that ssRNA is channeled through its multisubunit ring-like core into the active site tunnel of its exonuclease subunit Rrp44. Rrp44, both alone and in the exosome, processively cleaves RNA nucleotides one at a time, without consuming the energy of ATP, and releases a final 3-5 nucleotide product. We use molecular dynamics simulations and free energy calculations to identify the factors that enable processivity of RNA translocation and cleavage in the exosome complex. Our simulations reveal large and favorable free energies of RNA transfer from solution into the active site of Rrp44. The free energy profiles that characterize RNA translocation within the active site of Rrp44 are found to be dependent on the length of the RNA strand. While RNA strands formed by 5 nucleotides or more have downhill free energy profiles along the translocation coordinate towards the cleavage site, a 4-nucleotide RNA has a free energy barrier along the same coordinate, potentially leading to incomplete cleavage of ssRNA and the release of short (3-5) nucleotide products. Furthermore, dynamic insights gained from the performed simulations help elucidate the concerted nature of RNA translocation through the exosome complex. 2012-Pos Board B149 Probing DNA Bending Kinetics by yNhp6A with Ultrafast Temperature Jump Spectroscopy Manas K. Sarangi1, Molly Nelson-Holte2, Jim Maher2, Anjum Ansari1. 1 Department of Physics, University of Illinois at Chicago, Chicago, IL, USA, 2 Department of Biochemistry and Molecular Biology, Mayo Clinic College of Medicine, Rochester, MN, USA. The yeast Nhp6A protein is a member of the eukaryotic HMGB family of chromatin factors that enhance apparent DNA flexibility in many cellular processes. yNhp6A exhibits no sequence-specificity but binds to DNA with 1-10 nM affinity, sharply bending the DNA by >60 at the binding site. The kinetic mechanism by which this DNA deformation occurs remains unclear. It is not known whether the protein first binds weakly to unbent DNA and then deforms the DNA or if partially bent DNA conformations are thermally accessible and are ‘‘captured’’ and stabilized by the bound protein. The limited (tens-of-milliseconds) time resolution of previous kinetics studies was insufficient to observe the dynamics of DNA deformations in this complex. Here, we report time-resolved FRET measurements on yNhp6A bound to an 18-bp DNA oligomer labeled at each end with Cy3/Cy5, in response to a laser temperature-jump (T-jump) perturbation. The us temporal resolution of T-jump, together with ionic-strength and concentration dependence of equilibrium FRET and anisotropy measurements, helps reveal some of the microscopic kinetics steps. Equilibrium measurements with varied Nhp6A-DNA concentrations indicate that, at 250mM NaCl, the decrease in FRET with increasing temperature, in the range 15-60 C, is from unimolecular unbending of DNA. Kinetics traces measured under these salt conditions are singleexponential with relaxation times ranging from ~400 us (at 60 C) to ~1ms (at 45 C). At lower [salt] < 200mM NaCl, and at temperatures >65 C, biexponential kinetics are observed, likely corresponding to unimolecular bending/unbending, followed by dissociation of the complex and concurrent DNA melting. These results represent the first observation of DNA bending/ unbending dynamics in complex with a nonspecific DNA-binding protein, and are an important step towards a more comprehensive understanding of the kinetic mechanisms of DNA binding and bending interactions by this class of proteins. 2013-Pos Board B150 Single-Molecule DNA Melting Bubble Formation and Single-Strand Binding Protein Interaction Marko Swoboda, Lisa Hannusch, Maj Svea Grieb, Michael Schlierf. TU Dresden, Dresden, Germany. Exposed single-stranded DNA (ssDNA) in cells is threatened by degradation from nucleases (DNases) and prone to form secondary structures, which is why cells use single-strand binding proteins (SSB) to protect ssDNA. The most common example is exposure of the Okazaki fragments during replication, or double-stranded DNA melting induced by negative superhelicity, i.e. underwinding of double-stranded DNA caused by replication or transcription. Magnetic tweezers are the tool of choice to mimic and study the influence of superhelicity on DNA structure at the single-molecule level owing to the direct ability to apply torsional stress to double-stranded DNA. Here, we study the conditions of ssDNA exposure and formation from double-stranded DNA (dsDNA) samples with negatively supercoiled single DNA molecules with magnetic tweezers. Tuesday, February 10, 2014 Tracking double-stranded DNA extension with nanometer precision allows a direct observation of plectonemic coils formed during positive or negative supercoiling. Here, we use magnetic tweezers to set defined states of negative superhelicity with externally applied force. We can thus control the onset and behavior of DNA double strand separation (melting) and then vary parameters surrounding the formation of DNA melting bubbles. We find a strong SSB interaction with transient DNA bubbles even at low forces and superhelicities. SSB DNA bubble interaction is strongly force-dependent and additional superhelicity can displace SSB. 2014-Pos Board B151 Investigation of the Role Played by the RNA G-Quadruplex Structure in ALS/FTD Pathology Damian McAninch, Mihaela Rita Mihailescu. Chemistry and Biochemistry, Duquesne University, Pittsburgh, PA, USA. Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder resulting in motor neuron loss in brain and spinal cord. Frontotemporal dementia (FTD) is one of the most common forms of young onset dementia and second most common form of dementia overall, after Alzheimer’s, resulting in degeneration of temporal lobes along with personality changes and language impairment. ALS and FTD are now recognized as members of a broad continuum of neurodegenerative disorders, linked by similar pathology, mechanisms, and overlapping clinical symptoms. Two RNA-binding proteins of interest that link the two diseases are TAR DNA-binding protein 43 (TDP-43) and the fused in sarcoma/translocated in liposarcoma protein (FUS), which are the major protein components in over 90% of ALS and over 50% of FTD inclusions. We hypothesize that the G-quadruplex RNA structure might play an essential role in the pathogenic mechanisms of FUS in ALS and FTD. In this study, the G-quadruplex RNA binding properties of the wild type and C-terminal NLS mutant FUS protein implicated in ALS/FTD will be analyzed. 2015-Pos Board B152 Characterization of AIM2 DNA-Binding Properties and Filament Formation Seamus Morrone, Mariusz Matyszewski, Jungsan Sohn. Biophysics and Biophysical Chemistry, Johns Hopkins University, Baltimore, MD, USA. AIM2-Like Receptors (ALRs) are a class of innate immune receptors for foreign DNA, members of which exist in both the cytosol and nucleus. The eponymous member of this family, AIM2, is a cytosolic DNA sensor composed of an N-terminal pyrin domain (PYD) and a C-terminal HIN-200 domain (HIN). Previous studies of this protein has been limited to an N-terminal tagprotected version of the full-length or a truncated version of the protein. From these studies it has been concluded that AIM2 exists in a resting, autoinhibited form in which the PYD is bound to the HIN. Upon DNA binding, the PYD is released and forms a larger complex with its downstream partners to initiate the inflammation process. Recently, we have been successful in purifying the native (tag-less) full length protein. Using a combination of fluorescence, electron microscopy, and gel-shift assays, coupled with mutagenesis studies, we have characterized the DNA-binding properties of AIM2 and its ability to form filaments in solution. 2016-Pos Board B153 Characterization of IHF Binding to DNA Four-Way Junctions and Forks Veronica Birdsall, Vivian Deng, Ishita Mukerji. Wesleyan University, Middletown, CT, USA. Integration Host Factor (IHF) is an architectural protein that binds and bends DNA, facilitating the formation of protein-DNA complexes important for gene regulation. IHF binds with high affinity to a specific consensus sequence and induces a 160 bend upon binding. We have shown that IHF binds DNA four way junctions (4WJ) that do not contain the consensus sequence with nanomolar affinity and 1:1 stoichiometry for the specific interaction. We have also observed that IHF binds DNA forks with nanomolar affinity. The binding to junctions and forks is in direct contrast to IHF binding to linear duplex DNA, which is typically 1000-fold weaker. In this study we investigate whether the presence of the IHF consensus sequence influences IHF binding to DNA junctions and forks. We utilized gel shift and fluorescence binding assays to measure affinity and have observed that the high affinity for these nonnative structures is independent of the presence of the consensus sequence. We are further exploring how IHF binding influences junction conformation. Junction conformation is modulated by ion concentration where high concentrations of ions induces pairwise stacking of the helical arms resulting in quasi-continuous helices. In our investigations IHF binding to the nonconsensus junction induces an open conformation. We are specifically examining the distortion of the junction and DNA fork substrates upon IHF binding 401a using Fo¨rster Resonance Energy Transfer. Through these measurements, we are also exploring whether the mechanism of recognition differs between junctions and forks. 2017-Pos Board B154 Dynamics of Glyceraldehyde-3-Phosphate Dehydrogenase Interfacial Regions Affect Binding to AU-Rich RNA Michael White1, Mohsin Khan2, Daniel Deredge2, Christina Ross3, Royston Quintyn4, Beth Zucconi5, Vicki Wysocki4, Patrick Wintrode2, Gerald Wilson3, Elsa Garcin1. 1 Chemistry and Biochemistry, University of Maryland, Baltimore County, Baltimore, MD, USA, 2Chemistry and Biochemistry, University of Maryland, School, of Parmacy, Baltimore, MD, USA, 3Biochemistry and Molecular Biology, University of Maryland, School of Medicine, Baltimore, MD, USA, 4 Chemistry and Biochemistry, The Ohio State University, Columbus, OH, USA, 5Biochemistry and Molecular Biology, University of Maryland, Baltimore, Baltimore, MD, USA. The homotetrameric protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) has been shown to possess many functions aside from its role in glycolysis. Of particular interest is its role in post-transcriptional regulation. Despite lacking a canonical RNA binding motif, GAPDH has been shown to bind to many mRNAs and subsequently alter their translation. Most of these protein-nucleic acid interactions have been shown to occur by GAPDH binding to Adenine-Uridine Rich Elements (AREs) within the 3’ untranslated regions (UTRs) of specific mRNAs. While much evidence has been gathered in determining the means of RNA binding, the exact site and mechanism of binding still remain elusive. Variables that may be key to elucidating these two facets of RNA binding include the effects of posttranslational modifications, oligomerization, cofactor binding, and structural dynamics, of GAPDH. Herein, it is demonstrated for the first time that GAPDH binds to the core AREs of the tumor necrosis factor-a mRNA 30 UTR via a sequential two-step mechanism. As well, a single point mutation at the GAPDH dimer interface results in a reduction in binding affinity in the second step and an alteration in the bound RNA structure. In contrast to previous studies, it is shown here that this mutation does not affect protein oligomerization, but induces dynamic changes in protein regions localized along the P axis of the GAPDH tetramer. Based on our results, we propose a novel model for GAPDH binding to AREcontaining RNA that may be regulated by GAPDH post-translational modifications. Membrane Physical Chemistry II 2018-Pos Board B155 Calcium Effect on Directed Lipid Flow in Membrane: Improving Knowledge about Directed Cell Processes in Biological Cells Baharan Ali Doosti. Chalmers University of Technology, Gothenburg, Sweden. Observing the active role of lipids in response to chemical cues in artificial cell membranes could increase our understanding of directed cell transport phenomena in biological cells. Directed cell migration is essential in many biological processes including embryogenesis, wound healing, chronic inflammatory diseases, as well as cancer metastasis. Using biomimetic cell model systems makes it possible to use a minimal set of components for understanding directed cell movement and in-cell transport phenomena in regard to lipid sorting, formation of tubular protrusions and lipid movement. At present, we study directed lipid transport in artificial membranes by local biochemical gradient, calcium. We demonstrate that membrane tubulation and the flow of lipids in the membrane can be triggered and controlled by the chemical gradient applied along the lipid bilayer. This sheds light on interplay between membrane properties and chemical stimulation. 2019-Pos Board B156 Direct Measurement of Dipole Electric Field in Model Membranes using Vibrtaional Shifts of P-Cyanophenylalanine and Coupled with Molecular Dynamics Simulations Rebika Shrestha, Lauren J. Webb. Chemistry, University of Texas at Austin, Austin, TX, USA. The composition of biological lipid bilayer membrane creates a complex structural and electrostatic environment that regulates important membrane functions. The alignment of molecular dipole moments from lipid head groups and water molecules located at membrane-water interface creates the dipole potential (Vd). The dipole field (Fd) generated from this potential is the largest and about 1-10 MV/cm in magnitude, determined by indirect measurement techniques. It is located entirely within the membrane interior, and therefore, direct 402a Tuesday, February 10, 2014 measurement and manipulation of Fd is a challenge. Here, we present direct measurement of dipole electrostatic field within vesicular bilayer using vibrational Stark effect (VSE) shifts of nitrile oscillator in response to the local electrostatic field. We intercalated four different a-helix transmembrane peptides containing unnatural amino acid, p-cyanophenylalanine (p-CN-Phe) at four unique positions into unilamellar vesicles composed of 1,2-dimyristoyl-snglycero-3-phosphocholine (DMPC). Molecular Dynamics simulations of the membrane-intercalated helix containing two of the nitrile probes, one near the head-group region of the lipid and one buried in the interior of the bilayer were performed to examine the structure of nitrile with respect to the membrane normal, the assumed direction of Fd, by quantifying both tilt of the helix in bilayer and conformational rotation of the p-CN-Phe side chain at steadystate. As nitrile systematically moved towards the membrane interior, the vibrational absorption energies of nitrile showed blue shift. We used the measured VSE shifts and nitrile orientations within the membrane and calculated the magnitude of Fd to be 8 11 MV/cm, within the range reported in the literature. We increased the chemical and structural complexity of model membrane by adding cholesterol into vesicle composition at different concentrations and determined that Fd changes as function of membrane composition and temperature. 2020-Pos Board B157 Cell Penetrating Peptide Mediated Transport across Membranes Xin Li, Jing Huang, Matthew A. Holden. chemistry, UMass-Amherst, Amherst, MA, USA. We investigate how a small cell-penetrating peptide called Pep-1 is able to carry a large object, such as a protein, across a lipid bilayer. To create a membrane, two submicroliter, lipid-encased aqueous droplets are contacted termed a droplet interface bilayer (DIB). The peptides adsorb to the protein cargo non-covalently and somehow ‘‘carry’’ the protein from one droplet to the other through the membrane. We then quantitate the translocated cargo through a fluorogenic assay. We found that transport is dependent on voltage and membrane charge, and that the symmetry of the bilayer membrane may play a role in Pep-1-mediated protein translocation. Proteins as large as bgal (540 kDa) can be translocated using this method and we can detect as few as several thousand molecules. More recently, we’ve shown that trafficking can be monitored in real time, permitting a closer view of transport than ever before. We expect this new method will give some hints on the transportation driving force and make deciphering direct translocation mechanisms possible. 2021-Pos Board B158 Enhanced Membrane Permeability in E. coli Induced by Extracellular Adenosine Triphosphate Michael J. Wilhelm, Mohammad Sharifian Gh, Hai-Lung Dai. Department of Chemistry, Temple University, Philadelphia, PA, USA. It is well known that the addition of adenosine triphosphate (ATP) to the extracellular suspension of a variety of cell types increases the relative permeability of the cell membrane to a number of molecular species. We have previously demonstrated the use of time-resolved second-harmonic laser scattering (SHLS) and bright-field transmission microscopy (TM) as complementary real-time probes of molecular uptake in living cells (1,2) which are capable of revealing membrane-specific transport kinetics. Herein, we employ SHLS and TM to quantify the ATP-induced enhancement of membrane permeability in living Gram-negative bacteria. Specifically, we characterize the variation of the uptake rate of the cationic triphenyl methane dye malachite green (MG) into E. coli as a function of the concentration of ATP added to the extracellular suspension. As expected, due to the presence of the outer membrane (OM) porin channels, no change in the OM transport kinetics is observed. Conversely, transport across the cytoplasmic membrane (CM) is shown to increase by nearly an order of magnitude. Additionally, the presence of ATP results in an increase of MG adsorption onto the CM, suggesting that the permeability enhancement likely arises from a potential differential across the membrane. To gain mechanistic insight into the observed enhancement, we conduct control experiments using liposomes generated from lipids isolated from E. coli, including liposomes 1) composed solely of lipids, 2) with internalized ATP, 3) reconstituted with nucleoside transporters, and/or 4) reconstituted with ATP binding cassette cation transporters (and associated internalized cations). Subsequently, using model systems of gradually increasing complexity, the effectiveness of each membrane component on the observed permeability enhancement is compared. 1. Zeng, J., et al., (2013) Biophys. J., 104:139-145. 2. Wilhelm, M.J., et al., (2014) Chem. Phys. Lett., 605-606:158-163. 2022-Pos Board B159 Trans-Membrane Permeation Mechanism of Charged Methyl Guanidine Yukun Wang. Institute of Natural Sciences, Shanghai, China. ABSTRACT: The mechanism of trans-membrane ion permeation is studied using charged methyl guanidine as a model ion. With a widely applied reaction coordinate, our umbrella sampling results reveal a significant finite-size effect in small simulation systems and a serious hysteresis in large systems. Therefore, it is important to re-examine the simulation techniques for studying trans-membrane permeation mechanism of ions suggested in previous works. In this work, two novel collective variables are designed to acquire a continuous trajectory of the permeation process and small statistical errors through umbrella sampling. A water-bridge mechanism is discussed in detail. In this mechanism, a continuous water chain (or a chain of water molecules and lipid head groups) is formed across the membrane to conduct the trans-membrane permeation of charged methyl guanidine. We obtain a continuous transition trajectory by combining the two-dimensional umbrella sampling in the local region of the saddle state and a one-dimensional sampling in the out region. Our free energy analysis shows that, with the presence of the water-bridge, the energy barrier of the trans-membrane permeation of ions is reduced significantly. Our analysis suggests that the water-bridge mechanism is common for permeation of ions across thick membranes, including POPC and DPPC membranes. 2023-Pos Board B160 Imaging Potassium Flux through Individual Electropores Marc Szabo, Mark I. Wallace. Chemistry, University of Oxford, Oxford, United Kingdom. Imaging the ion flux through channels or membrane pores using fluorescent indicator dyes is an attractive alternative to patch-clamping as a means to make parallel measurements from individual channels. Current methods are restricted to imaging calcium-permeant channels, primarily due to the limitations in dye performance. Here we present a method for potassium flux imaging using TIRF microscopy in droplet interface bilayers. Using the potassium indicator dye APG-4 we visualize the formation of individual electropores, and examine the response of pores to the applied potential. 2024-Pos Board B161 Variable Adhesion Strength for Giant Unilamellar Vesicles Controlled by External Electrostatic Potentials Jan Steinkuehler, Jaime Agudo-Canalejo, Reinhard Lipowsky, Rumiana Dimova. Theory & Bio-Systems, Max Planck Institute of Colloids and Interfaces, Potsdam, Germany, Potsdam, Germany. We developed an experimental setup to study adhesion of giant unilamellar vesicles (GUVs) with variable adhesion strength. The vesicles adhere to a planar substrate. The adhesion is easily modulated by the application of an external potential to transparent ITO electrodes (the substrate) which enables us to study single GUVs during adhesion. The adhesion energy is calculated from the vesicle shape assessed with confocal microscopy. We implement a theoretical method for deducing the adhesion energy based on the overall vesicle shape. The results show that for the explored ranges of the external potential, the adhered vesicles are in the weak adhesion regime. Using fluorescence quenching assays, we conclude that the outer membrane leaflet is depleted of the negatively charged dye NDB-PG in the adhesion zone. Based on these results, we discuss the driving forces involved in the vesicle adhesion. Using FRAP measurements, we also show that diffusion is only slightly hindered in the adhesion zone. Some first results on applying our adhesion approach to study demixing in multicomponent membranes will be presented. We will demonstrate that adhesion can be used to modulate the phase state in the adhering and nonadhering regions of vesicles exhibiting coexistence of liquid ordered and liquid disordered domains. 2025-Pos Board B162 Single Cell Force Spectroscopy Analysis for Acinetobacter Baylyi Mutation Aggregation Mehrdad M. Tajkarimi1, Albert M. Hung2, Scott H. Harrison3, Jeffrey E. Barrick4, Joseph L. Graves, Jr2. 1 Physical sciecne, Forsyth Tech, Winston-Salem,, NC, USA, 2 Nanoengineering, North Carolina A&T State University, Greensboro, NC, USA, 3Biological science, North Carolina A&T State University, Greensboro, NC, USA, 4Molecular Biosciences, Institute for Cellular and Molecular Biology , University of Texas Austin, Austin, TX, USA. Acinetobacter baylyi ADP1 is an ideal bacterial strain for synthetic biology and metabolic engineering due to its high natural transformability and capability for Tuesday, February 10, 2014 processing diverse chemical compounds. For the production of compounds within industrial bioreactors, it is optimal to have a uniform dispersal of bacterial cells within solution, but mutations in bacterial cells may generate surface properties leading to cell aggregation. During long-term culture in the laboratory, mutants of the bacterium A. baylyi ADP1 originated a distinctive phenotype of cell aggregation. Genome sequencing and the analysis of gene knockouts showed this aggregation to be due to mutations in the per and pgi genes, and a reduction in bioemulsifier production. Qualitative analysis of Atomic Force Microscopy (AFM) visualizations identified altered appearances of cell surfaces correlating with the difference in cell aggregation phenotype. AFM force spectroscopy experiments were then conducted to compare the adhesive and viscoelastic properties of aggregating cells to non-aggregating cells. The most distinctive difference found for force spectroscopy measurements was for a four-fold difference in nN in adhesion that was attributable to pgi. Overall, this experiment has resulted in a multilevel approach for the evaluation and detection of a cell aggregation phenotype in mutant strains of A. baylyi ADP1. 2026-Pos Board B163 Membrane Environment can Enhance the Interaction of Glycan Binding Protein to Cell Surface Glycan Receptors Lei Shen1, Yini Wang2, Chia-I Lin3, Hung-wen Liu3, Athena Guo2, Xiaoyang Zhu1. 1 Department of Chemistry, Columbia University, New York, NY, USA, 2 MicroSurfaces, Inc., Englewood, NJ, USA, 3College of Pharmacy, University of Texas at Austin, Austin, TX, USA. The binding of lectins to glycan receptors on the host cell surface is a key step contributing to the virulence and species specificity of most viruses. This is exemplified by the viral protein hemagglutinin (HA) of the influenza A virus, whose binding specificity is modulated by the linkage pattern of terminal sialic acids on glycan receptors of host epithelial cells. Such specificity dictates whether transmission is confined to a particular animal species or jumps between species. Here we show, using H5N1 avian influenza as a model, that the specific binding of recombinant HA to a2-3 linked sialic acids can be enhanced dramatically by interaction with the surface of the lipid membrane. This effect can be quantitatively accounted for by a two-stage process in which weak association of HA with the membrane surface precedes more specific and tighter binding to the glycan receptor. The weak protein-membrane interaction discovered here in the model system may play an important secondary role in the infection and pathogenesis of the influenza A virus. 2027-Pos Board B164 Impact of Composition upon Ordered Membrane Domain(‘‘Raft’’) Formation by Lipids from Pathogenic Bacteria Zhen Huang. Biochemistry and Cell Biology, Stony Brook University, Stony Brook, NY, USA. Co-existing ordered (raft) and disordered membrane domains have been indentified in the outer membrane of the pathogenic bacterium Borrelia burgdorferi, the bacterium which causes Lyme disease. Co-existing ordered and disordered membranes can also be detected into B. burgdorferi lipid extracts. However, unlike eukaryotic cells, B. burgdorferi lack sphingolipids, which are crucial component of eukaryotic rafts. In order to understand the basis of domain formation in this organism we have isolated the major lipids of B. burgdorferi by thin layer chromatography, and have initiated studies of their physical properties when dispersed in aqueous solutions. We have found that mixtures of the predominant lipids found in B. burgdorferi, namely, ACGal, a lipid in which a fatty acyl chain and cholesterol are linked to galactose, monogalactosyldiglyceride (MGalD) with phosphatidylcholine (PC) can form ordered domains with thermal stabilities similar to that in whole lipid extracts. However, for individual lipid aqueous dispersions domain formation and/or stability is very different than in whole lipid extracts. Combinations of B. burgdorferi lipids are being studied to identify which lipid are necessary and sufficient for the formation of co-existing ordered and disordered domains in this bacterium and related ones. 2028-Pos Board B165 Stabilization of Glycosphingolipid Domains by Palmitoyl Ceramide in Unsaturated Phosphatidylcholine Bilayers Md. Abdullah Al Sazzad, J. Peter Slotte, Max Lo¨nnfors. ˚ bo Akademi University, Turku, Finland. Biochemistry, Dept of Bioscience, A Ceramides and glycosphingolipids (GSLs) are minor components in most eukaryotic cells. Since ceramides may be generated in lipid raft like domains by enzyme degradation of sphingomyelin (SM), ceramide/GSL interactions may become relevant in cell membranes. To examine their mutual interactions, we have prepared binary and ternary model bilayer systems composed of a 403a disordered lipid (unsaturated phosphatidylcholine), and different combinations of saturated sphingolipids (palmitoyl SM, palmitoyl ceramide (PCer), and hydroxylated or non-hydroxylated galactosyl or glucosyl palmitoyl-ceramide (PGalCer or PGlcCer)). We have used trans-parinaric acid (tPA) as a probe to detect the ordered domains formed by the sphingolipids in the phosphatidylcholine bilayer. In binary systems, the PCer formed the most thermostable ordered domains, followed by PGalCer, OH-PGalCer, OH-PGlcCer, and PGlcCer. The PSM domains were the least thermostable. Addition of PCer to the GSL or PSM domains increased their thermostability, with the exception of PGalCer, whose thermostability was unaffected by inclusion of PCer. Lifetime analysis of tPA suggested that all sphingolipid ordered domains became even more ordered in the presence of PCer. We conclude that PCer was able to interact with all the examined sphingolipids and increased packing order in the domains. 2029-Pos Board B166 Comparison of Line Tension Measurement Techniques in Phase Separated Multi-Component Lipid Monolayers Juan TigreLazo1, Joan C. Kunz2, Vision Bagonza3, Andrew H. Nguyen1, Emil Eldo2, Benjamin L. Stottrup1. 1 Physics, Augsburg College, Minneapolis, MN, USA, 2Chemistry, Augsburg College, Minneapolis, MN, USA, 3Biology, Augsburg College, Minneapolis, MN, USA. Langmuir monolayers of multi-component lipid compositions have been used to study the mixing behavior of sterol-phospholipid systems. Using traditional Langmuir pressure-area isotherms and fluorescence microscopy techniques we compare line tension measurements using two methods of image analysis. Line tension between coexisting phases of sterol-rich and sterol-poor domains can be extracted from a Fourier analysis of domain boundary fluctuations (J. Phys. Chem. B, 111:11091-11094). These measurements will be compared to a recently developed non-perturbative technique based on domain size distribution (Proc. Natl. Acad. Sci. 110:13272-1327). Until now these two measurement techniques have not been compared on the same data set. The compositions studied include 30:70 mixtures of cholesterol and DMPC, DLPC, and DCPC. As well as 25:75 mixtures of 25-hydroxycholesterol DMPC systems. 2030-Pos Board B167 The Average Area Per Molecule of Cholesterol/PC-Lipid Bilayers: A Review of Experimental Data and a Physically Inspired Model Jonathan P. Litz, Sarah L. Keller. Chemistry, University of Washington, Seattle, WA, USA. We recently documented that beta-cyclodextrin extracts cholesterol at different rates from supported lipid bilayers containing either DMPC, SOPC, or DOPC [Litz & Keller, BJ, 2013, 93A]. Quantitative measurement of the rate of cholesterol depletion relies on accurate knowledge of the average area per molecule within each bilayer, as does calibration of fixed-area molecular dynamic simulations [e.g. Klauda & Nagle, BJ, 2006, 2796]. A challenge is to integrate a plethora of seemingly incompatible experimental results, which yield significantly different average areas per molecule of PC-lipid/cholesterol bilayers. Historically, disagreements between values derived from x-ray and neutron scattering have been attributed to differences in sensitivity between the two techniques, and more recent approaches have analyzed scattering data from both techniques [e.g. Kucerka & Katsaras, BJ, 2008, 2356]. Here I show that the majority of the data from which areal measurements are derived is in agreement, and that most disparity in reported values arises from the choice of difficult-to-measure physical parameters. I provide an estimate of the uncertainty of how the area of a PC-lipid bilayer changes as a function of the mole fraction of cholesterol and derive a physically-inspired, two-parameter model to predict the change. I compare the efficacy of my model with that of the currently preferred four-parameter model [Edholm & Nagle, BJ, 2005, 1827]. I then apply my results to quantitatively report rates of cholesterol depletion from two-component lipid bilayers. 2031-Pos Board B168 Cholesterol Bilayer Domain in Phospholipid Bilayer Membranes can be Detected by Confocal Microscope Marija Raguz1, Nada Ilic2, Suresh Kumar3, Mariusz Zereba4, Laxman Mainali5, Witold K. Subczynski5. 1 Medical Physics and Biophysics, University of Split, Split, Croatia, 2 Physics, University of Split, Split, Croatia, 3Pathology, Medical College of Wisconsin, Milwaukee, WI, USA, 4Ophthalmology, Medical College of Wisconsin, Milwaukee, WI, USA, 5Biophysics, Medical College of Wisconsin, Milwaukee, WI, USA. The unique feature of the eye lens fiber-cell plasma membrane is its extremely high cholesterol content; cholesterol/phospholipid molar ratio can be as high as 404a Tuesday, February 10, 2014 4 in human lens nucleus. Cholesterol saturates bulk phospholipid bilayers and induces formation of immiscible cholesterol bilayer domains (CBDs) within membranes. The presence of CBDs plays crucial role ensuring that surrounding phospholipid bilayers are saturated with cholesterol which keeps the bulk physical properties of lens-lipid membranes consistent and independent of changes in phospholipid composition. Thus, CBDs help to maintain lens-membrane homeostasis when the membrane phospholipid composition changes significantly with age. Our previous experiments were based on electron paramagnetic resonance spin-labeling methods. They allowed to detect CBDs in model and lens lipid membranes at high cholesterol content (1,2). In current experiments the giant unilamellar vesicles made of cholesterol/distearoylphosphatidylcholine (Chol/DSPC) mixtures, with mixing ration from 0.5 to 5 and labeled with phospholipid analog (1,10 -Dioctadecyl-3,3,3’,30 -Tetramethylindocarbocyanine5,50 -Disulfonic Acid) and cholesterol analog (23-(dipyrrometheneboron difluoride)-24-norcholesterol) fluorescent probes, were prepared using the electroformation method (3). Confocal microscopy experiment allowed us to confirm that membranes with higher Chol/DSPC molar ratio contain two distinct lipid environments, the bulk phospholipid-cholesterol domain (PCD) and the CBD. The amount of CBD was greater in membranes with higher Chol/DSPC ratio. According to these results, CBD is always located on the top of the giant vesicle. Rationale for this discovery is that CBD has smaller mass per unit area of the vesicle compared with PCD. Additionally, it can be concluded that vesicles with higher Chol/DSPC molar ratio are more rigid and show less wobbling motion in water environment. 1. Raguz et al. CPL 2011 164(8):819-29. 2. Mainali et al. BBA 2013 1828(6):1432-40. 3. Veatch SL. Methods Mol Biol. 2007 398:59-72. Acknowledgements Supported by NIH grants EY015526 and EY001931. 2032-Pos Board B169 Correlated Motion and Complex Formation of Lipid-Raft Components Analyzed by High-Resolution Secondary Ion Mass Spectrometry Monica M. Lozano1, Jennifer S. Hovis1, Frank R. Moss III1, Krishna Kumar2, Steven G. Boxer1. 1 Department of Chemistry, Stanford University, Stanford, CA, USA, 2 Department of Chemistry, Tufts University, Medford, MA, USA. It is widely believed that certain membrane lipids and membrane-anchored proteins associate to form clusters with emergent function. While extensive data on phase diagrams are available for a range of lipid compositions, these are most commonly visualized by the partitioning of dyes between different phases. We have been developing the use of secondary ion mass spectrometry imaging to directly visualize molecules of interest within supported lipid bilayers (Lozano et al. JACS 2013). Specifically, we want to address the question which neutral membrane components associate with the well-known lipid-raft marker ganglioside GM1, which has a single negative charge, after GM1 is reorganized by an in-plane electric field on a patterned supported bilayer (SLB). NanoSIMS imaging was used to generate molecule-specific concentration profiles of several SLB compositions following electrophoresis: GM1/DOPC, GM1/CHOL/ DOPC, and GM1/CHOL/PSM/DOPC. In a control SLB sample without GM1 it is observed that CHOL, PSM, and DOPC do not reorganize by membrane electrophoresis. However, NanoSIMS images clearly illustrate that PSM and CHOL associate with GM1 as it is transported toward the positive electrode while DOPC is displaced in the opposite direction towards the negative electrode. Further analysis of the concentration profiles for the co-diffusing components (GM1/CHOL/PSM) suggest the formation of a 4:2:1 PSM: GM1:CHOL stoichiometric complex. These results demonstrate that both cholesterol and sphingomyelin do tend to associate with GM1 and we might speculate that similar behavior would be observed for GPI or myristolyated proteins. 2033-Pos Board B170 Domain Morphologies of Complex Phosphoinositide/Lipid Langmuir Films in the Presence of Bivalent Cations Katrice E. King, Arne Gericke. Chemistry and Biochemistry, Worcester Polytechnic Institute, Worcester, MA, USA. Although phosphatidylinositol (PI) and phosphoinositides (PIPs) only comprise a small percentage of the inner leaflet of the plasma membrane, they mediate a large variety of signaling events. In previous studies, we have observed the absence of macroscopically discernible domains in mixtures of PI/PC and PI(4,5)P2/PC. The addition of cholesterol to these mixtures results in condensation of the monolayer and hence domain formation. To better mimic the ionic conditions and hydrogen bonding properties of the inner leaflet plasma membrane, we investigated in this study the effect of common inner leaflet plasma membrane lipids like phosphatidylethanolamine (PE), phosphatidylserine (PS) and PI, on phosphoinositide domain behavior in the presence of cholesterol and/or bivalent cations. We find that the addition of varying concentrations of Ca2þ to PI(4,5)P2/cholesterol monolayer leads to a size reduction of the domains. We hypothesize that this is due to a penetration of the Ca2þ ions into the PI(4,5)P2 headgroup region, leading to a disruption of the hydrogen bond network formed by the PI(4,5)P2 headgroup and cholesterol. We find for PE/ PI(4,5)P2 and PI/PI(4,5)P2 homogeneous mixing of the monolayer. The addition of cholesterol to PE/PI(4,5)P2 leads to the formation of small domains at low surface pressures that disappear at higher pressures. In addition to cholesterol, we also investigate in this study the effect of bivalent cations on these lipid mixtures. 2034-Pos Board B171 Ceramide and Cholesterol Effects on Phospholipid Bilayers under the AFM: Characterization of Complex Lipid Phases Aritz B. Garcı´a-Arribas, Jon V. Busto, Alicia Alonso, Felix M. Goni. Unidad de Biofisica (CSIC-UPV/EHU), Leioa, Spain. Atomic force microscopy (AFM) has been applied to the characterization of palmitoylceramide (pCer) and cholesterol (Chol) incorporation into phospholipid-based supported planar bilayers (SPBs) at 22 C. Phospholipids were dipalmitoyl phosphatidylcholine (DPPC) or palmitoyl sphingomyelin (pSM). Membranes of different compositions were prepared by either the vesicle adsorption or the spin-coating procedures (the latter strictly for pCercontaining mixtures) and analyzed with a combination of AFM imaging (domain segregation, bilayer thickness and roughness) and force spectroscopy (mechanical resistance to indentation). The mixtures under study were pure phospholipids (pSM, DPPC), phospholipid:Chol (70:30), phospholipid:Chol:pCer (2:1:1) and phospholipid:pCer (90:10, 80:20 and 70:30). Binary phospholipid:pCer mixtures at increasing ceramide ratios gave rise to highlyresistant segregated domains with increasing extension but similar properties in terms of breakthrough forces, thicknesses and roughnesses. These ceramide-enriched domains are able to exclude a fluorescent lipid probe (DiIC18) due to their high intermolecular packing. Interestingly, these domains have been reported to disappear when model membranes become highly enriched in cholesterol in fluid membranes or in the absence of a fluid phase (our case). Indeed, the ternary mixtures (2:1:1) gave rise to a homogenous lamellar gel phase with significantly different properties when compared to all of the other mixtures under study: ternary mixtures showed a reduced thickness and an intermediate roughness and mechanical resistance when compared to phospholipid:Chol (70:30) and phospholipid:pCer. These differences were statistically significant. More importantly, at those relatively high pCer and Chol concentrations in ternary mixtures, no mutual displacement of these molecules was observed. The data become relevant in the context of sphingolipid signaling and membrane platform formation. 2035-Pos Board B172 End-Product Diacylglycerol Enhances Activity of Phosphatidylinositol Phospholipase C through Changes in Membrane Lipid Domain Structure Hasna Ahyayauch1,2, Jesu´s Sot1, M. Isabel Collado3, Nerea Huarte1, Jose´ Requejo-Isidro1, Fe´lix M. Gon˜i1, Alicia Alonso1. 1 Unidad de Biofisica (CSIC-UPV/EHU), Leioa, Spain, 2Institut Supe´rieur des Professions Infirmie`res et des Techniques de Sante´, Rabat, Morocco, 3SGiker, University of the Basque Country, Leioa, Spain. DAG-induced activation of PI-PLC has been studied using as substrates vesicles containing PI, either pure or in mixtures with DMPC, DSPC, sphingomyelin, or galactosylceramide. At 22 C DAG at 33 mol% increases PI-PLC activity in all the mixtures, but not in pure PI bilayers. DAG also causes an overall decrease in DPH polarization (decreased molecular order) in all samples, and increased overall enzyme binding. Confocal fluorescence microscopy examination of GUV of all the compositions under study, with or without DAG, and quantitative evaluation of the phase behaviour using LAURDAN generalized polarization, and of enzyme binding to the various domains, indicate that DAG activates PI-PLC whenever it can generate fluid domains to which the enzyme can bind with high affinity. In the specific case of PI:DMPC bilayers at 22 C DAG induced increased enzyme binding and activation, but no microscopic domain separation was observed, the presence of DAG-generated nanodomains is proposed instead for this system. In PI:galactosylceramide mixtures DAG may exert its activation role through the generation of small vesicles, that PI-PLC is known to degrade at higher rates. In general our results indicate that global measurements using fluorescent probes in vesicle suspensions in cuvette are not enough to understand DAG effects that take place at the domain level. The above data reinforce the idea of DAG as an important physical agent regulating membrane or cell properties. Tuesday, February 10, 2014 2036-Pos Board B173 Lessons from Kinetics: Assessing Nuances in Bilayer Properties by Examining Equilibration John D. Bell, Clinton S. McCleskey, Joseph Chen, Emma R. Moulton, Morgan M. Schwab, Holli K. Wiberg. Phyiology and Developmental Biology, Brigham Young University, Provo, UT, USA. The equilibration of five fluorescent probes was compared for bilayers composed of dipalmitoylphosphatidylcholine or dipalmitoylphosphatidylglycerol using pH and temperature as independent variables. The probes used were diphenylhexatriene and its trimethylammonium derivative (DPH and TMA-DPH), Laurdan, Patman, and F2N12S. The equilibration kinetics involved multiple exponentials and both positive and negative changes in emission intensity. The kinetics of the equilibration process depends on the relative contributions of four probe characteristics: 1) probe water solubility, 2) charge barriers to probe insertion, 3) photobleaching, 4) effect of the probe on the local membrane environment. The initial rates of DPH and TMA-DPH equilibration appeared related to their relative water solubility. Subsequent decays in emission intensity observed for both probes were explained by photobleaching. Interestingly, the photobleaching rate for TMA-DPH was influenced by lipid structure and phase. Additional changes in membrane structure following probe insertion were implied by slow increases in emission intensity. Patman differed from the other probes in that membrane surface charge strongly limited its ability to insert into the bilayer. Patman and Laurdan fluorescence at dual wavelengths provided additional evidence for slow local membrane changes responding to the presence of the probe. These changes varied with both membrane phase and charge. Finally, the initial rate of F2N12S equilibration was very rapid, probably due to its high water solubility. Latent changes in the intensity and emission maximum of F2N12S also argued for slow local membrane alterations provoked by the probe. In this case, the latent changes often included decreases in intensity that were not caused by photobleaching. Although most of these differences among the probes can be rationalized based on probe structure, the additional slow changes to local membrane environment following probe insertion appear to be a property of membrane structure. 2037-Pos Board B174 The Chemical Potential of Cholesterol Regulates the Pro-Metastatic Phenotype in a Cell Culture Model of Breast Cancer Artem G. Ayuyan, Fredric S. Cohen. Molecular Biophysics and Physiology, Rush University Medical Center, Chicago, IL, USA. We have developed a simple and reliable method to determine the chemical potential of cholesterol in plasma membranes of living cells in vitro. For a variety of cultured cells, including non-metastatic breast cancer cells, the chemical potential of cholesterol is maintained at a level of about 1.9 kBT per moleculerelative to crystalline cholesterol. But for a metastatic breast cancer cell line, this chemical potential is appreciably greater, at about 0.7 kBT per molecule. In light of these observations and recent reports of cholesterol’s role in breast cancer progression we have developed a method to alter and maintain the chemical potential of cholesterol in plasma membranes of cultured cells at any desired level. Through this technique, we lowered the chemical potential of cholesterol in the metastatic cell line to that of the non-metastatic line and found that this significantly reduced the expression levels of several key proteins implicated in breast cancer progression and metastatic spread. 2038-Pos Board B175 Membrane Resistance to Detergent-Induced Solubilization as a Matter of Physical Phase in Binary Lipid Mixtures Bruno Mattei, Ana David, Cruz Franc¸a, Karin Amaral Riske. Biophysics, UNIFESP, Sa˜o Paulo, Brazil. Molecular biology protocols routinely use detergents for extraction of membrane or intracellular components. However, detergent-induced solubilization usually results in the presence of membrane debris, with similar composition to membrane rafts and liquid-ordered (Lo) phase. In the present work we have studied the solubilization by the detergent Triton X-100 (TX-100) of membranes composed of different lipid mixtures and phases. The membrane compositions evaluated were palmitoyl-oleoyl-phosphatidylcholine (POPC) and POPC:cholesterol (7:3) (liquid disordered phase, Ld), Sphingomyelin (SM, Gel phase) and SM:cholesterol (7:3) (Lo phase). The solubilization was followed by optical microscopy of giant unilamellar vesicles (GUVs), showing that POPC and SM membranes are totally solubilized by TX-100, while POPC:cholesterol membranes are partially solubilized and SM:cholesterol membranes are completely insoluble. The incorporation of TX-100 in GUVs was measured trough area increase for vesicles in fluid phases, showing that for POPC membranes, the addition of cholesterol diminishes the incorporation 405a of TX-100 and for vesicles of SM:cholesterol the area increase very low. Light scattering of large unilamellar vesicles (LUVs) titrated with TX-100 confirmed the results of optical microscopy. The isothermal titration calorimetry (ITC) of LUVs with detergent showed that the addition of cholesterol diminishes the partition coefficient of TX-100 in both POPC and SM membranes. In addition, enthalpic changes are observed in the same detergent-lipid molar ratios associated with the onset and completion of the solubilization process. Differential scanning calorimetry of SM LUVs with detergent show detergent-induced fluidification of the gel phase, which could account for part of the large endothermic contribution measured with ITC. Our result suggests that cholesterol increases membrane resistance to detergent-induced solubilization and confirmed that the Lo phase is virtually insoluble. Supported by: FAPESP and CNPq Membrane Fusion 2039-Pos Board B176 Actin and Dynamin Control the Fate of the Fusion Intermediate - the g-Profile Peter Wen. NINDS, NIH, bethesda, MD, USA. Fusion generates an intermediate structure, an U-shaped membrane profile, to release vesicular contents. The subsequent fate of the intermediate structure, either U-profile pore closure or merge between the U-profile and the plasma membrane, determines whether retrieval of the U-profile is via fusion pore closure or classical endocytosis. Owing to difficulty of detecting U-profile, the mechanism controlling the U-profile’s fate, particularly the merging process, is entirely unclear, and is often assumed an automatic process without using energy or force. By directly imaging U-profile in neuroendocrine chromaffin cells, we found that Uprofile merging requires ATP hydrolysis and is mediated by actin dynamics that provides a mechanical force to shrink the U-profile, whereas dynamin counteracts vesicle merging by mediating Uprofile closure, which precludes U-profile merging. These results reveal actin, ATP, and dynamin in control of the fate of the fusion intermediate and thus the endocytosis route for recycling of fusing vesicles. 2040-Pos Board B177 The Molecular Mechanism of Monolayer Scission Shachi Katira1, Berend Smit2. 1 Bioengineering, University of California, Berkeley, Berkeley, CA, USA, 2 Chemical and Biomolecular Engineering, University of California, Berkeley, Berkeley, CA, USA. Hydrophobic globules surrounded by lipid monolayers are ubiquitous structures found in most cells, prokaryotic and eukaryotic. These structures, called lipid droplets, are hypothesized to accumulate as inclusions of oil-like molecules between the two leaflets of a membrane which grow larger and eventually bud off. The budding process is submicroscopic and difficult to observe in experiments, and studying it in molecular simulations has thus far presented computational challenges. Using dissipative particle dynamics, we attempt to understand the molecular details of the droplet budding process. We develop a lipid reservoir that can supply lipids to the bulging monolayer as the droplet buds out of the membrane. Our simulations support existing schematic models for the growth and budding process and predict a morphological transition between a partially and completely enveloped intermediate structure. Our results suggest a molecular mechanism for monolayer scission. Droplets generated using this technique can be useful in further studying the structure and dynamics of the droplet and its unique monolayer-integrated proteome. 2041-Pos Board B178 Lipid Transfer Kinetics from Nanolipoprotein Particles to Bicelles Robert Renthal1, Ginny Lai1, Kevin Munoz Forti2. 1 Biology, University of Texas at San Antonio, San Antonio, TX, USA, 2 Biology, University of Puerto Rico, Ponce, PR, USA. Nanolipoprotein particles (NLPs), also known as nanodiscs, are lipid bilayers bounded by apolipoprotein. We recently showed that lipids and membrane proteins cannot exchange between NLPs. However, addition of bicelles opens NLPs and transfers their contents to bicelles, which freely exchange lipids and proteins. Because monomeric membrane proteins can be prepared in NLPs by cell-free protein synthesis, the bicelle-induced transfer process may provide a new method for studying membrane protein oligomerization. The mechanism of the NLP-bicelle interaction is unknown. We have now tested the effects of bicelle detergent (DHPC), apolipoprotein (MSP1E3D1), and temperature on lipid transfer from NLPs to bicelles, using stopped-flow kinetics. NLPs were prepared with fluorescent lipids (0.02 to 0.05 mole fraction), consisting of FRET donors (NBD-PE) and acceptors (LR-PE) at approximately 406a Tuesday, February 10, 2014 equal concentrations. NLPs were mixed with a 200-fold molar excess of DHPC/DMPC bicelles (equimolar 6- and 14-carbon acyl chains) in a stopped-flow fluorometer. The rate of lipid transfer was monitored by the appearance of unquenched NBD fluorescence at 520 nm. The observed pseudo-first-order rate constant was surprisingly small (0.26/sec). NLPs did not react with DHPC alone below its critical micelle concentration (cmc). Above the cmc, the reaction was complete within the instrument dead time. Thus, the rate-limiting step is not the reaction of NLPs with DHPC monomers or micelles. Added MSP1E3D1 had no effect on the rate, ruling out free apolipoprotein involvement. The NLP-bicelle mixing rate showed a strong temperature dependence (activation energy ¼ 28 kcal/mol). Near or below the DMPC phase transition temperature, the kinetics were biphasic. The results suggest NLP-bicelle mixing kinetics may be mechanistically similar to lipid mixing via fusion pores. 2042-Pos Board B179 Drunken Membranes: How does Ethanol Impact Fusion of Vesicles to Planar Lipid Bilayers? Brady Hunt1, Jason R. Paxman1, D. Coulson Huntington2, Dixon J. Woodbury1. 1 Physiology and Developmental Biology, Brigham Young University, Provo, UT, USA, 2Physics and Astronomy, Brigham Young University, Provo, UT, USA. Previously we have shown that fusion of vesicles to a membrane is changed by lipid phase as altered by cholesterol and temperature (Lee et al. 2013, Chem Phys Lipids 166:45-54). Ethanol is also known to alter membrane phase behavior. To investigate the effect of ethanol on fusion rate, we used the Nystatin/Ergosterol (NYS/ERG) fusion assay. We measured fusions per minute using planar membranes containing PE/PC/CHL and vesicles containing PE/PC/PS/ERG. For our initial experiments we used high concentrations of ethanol added to either side of the bilayer. At 1% ethanol an increase was observed, and at 4% fusion rates increased 3 fold compared to control when added to the cis side of the bilayer (same side as vesicles). However, no significant increase was observed when added to the trans side. Similar results were observed with methanol. It also appears that the 2-3 fold increase in fusion rate occurs regardless of whether ethanol is added before or after vesicles. We hypothesize that this effect of ethanol is to alter lipid phase behavior of the vesicle membrane. To verify the effects of ethanol on membranes we utilized differential scanning calorimetry (nDSC). Solutions of KCl buffer and DPPC vesicles were analyzed with and without ethanol. The DPPC vesicles with ethanol were shown to have a transition state ~0.5 C lower than without ethanol. This is consistent with our hypothesis that adding ethanol alters lipid fluidity and/or phase behavior. 2043-Pos Board B180 Role of Electrostatic Interactions in the Anchoring of Dengue E Protein to Lipid Membranes Juan M. Vanegas1, David M. Rogers2, Michael S. Kent1, Susan B. Rempe1. 1 Center for Biological and Materials Sciences, Sandia National Laboratories, Albuquerque, NM, USA, 2Department of Chemistry, University of South Florida, Tampa, FL, USA. Infection of host cells by Dengue virus occurs in the late endosome, where the viral envelope protein, E, mediates the fusion of the viral and endosomal membranes. The low pH triggers a conformational change that first causes the tip of E to project out from the surface of the virus and bind the endosomal membrane. E forms trimers during this process and further conformational changes in the protein force the viral and endosomal membranes into contact. Binding of E to the endosomal membrane has been largely attributed to the hydrophobic fusion loop at the tip of E, as several residues in this region are highly conserved. However, fusion requires anionic lipids in the endosomal membrane, suggesting that binding of E may also be modulated by electrostatic interactions. Here, we use atomistic molecular dynamics simulations to study the interaction between Dengue E trimer and various lipid membranes composed of POPE, POPC, POPG (anionic), and cholesterol. We use a truncated E trimer consisting of roughly 1/3 of the residues (those near the tip). Truncated E retains the structural features of the full protein and significantly reduces the computational costs. We find that E inserts into all membranes tested regardless of the presence of anionic lipids or cholesterol. During insertion, the dominant interactions occur hydrogen bonds between the positively charged lysines and oxygens of the lipid headgroups. These interactions induce tilting of the protein that is accompanied by a large deformation of the membrane. This local deformation of the membrane (inducing negative curvature) produced by electrostatic interactions may play an important role during fusion. The simulations may also suggest that the hydrophobic residues in the fusion loop make only a small contribution to the binding energy. 2044-Pos Board B181 Calcium Sensitive Ring-Like Oligomers of Synaptotagmin: Implications for Regulation of Neurotransmitter Release Shyam Krishnakumar. Dept of Cell Biology, Yale University, New Haven, CT, USA. The synaptic vesicle protein synaptotagmin-1 (SYT) is required to couple calcium influx to the membrane fusion machinery. However, the structural mechanism underlying this process is unclear. Using electron microscopy we find an unexpected circular arrangement (ring) of SYT’s cytosolic domain (C2AB) formed on lipid monolayers in the absence of free calcium. Rings vary in diameter from 18 nm to 43 nm, corresponding to 11 to 26 molecules of SYT. Continuous stacking of the SYT rings occasionally converts both lipid monolayers and liposomal bilayers into protein-coated tubes. Helical reconstruction of the SYT tubes shows that the C2B interacts with the membrane and is involved in ring formation, while the C2A domain points radially outwards. SYT rings are rapidly disrupted by physiological concentrations of free calcium but not by magnesium. Assuming that calcium-free SYT rings are physiologically relevant, these results suggest a simple and novel mechanism by which SYT regulates neurotransmitter release: the ring acts as a spacer to prevent the completion of SNARE complex assembly thereby clamping fusion (in the absence of calcium). When the ring disassembles in the presence of calcium, then fusion proceeds unimpeded. 2045-Pos Board B182 Viral Membrane Fusion at Single Pore Resolution Brett E. Alcott1,2, Zhenyong Wu2,3, Ben O’shaughnessy4, Erdem Karatekin2,3. 1 Biochemistry and Molecular Biophysics, Columbia University, New York, NY, USA, 2Cellular and Molecular Physiology, Yale University, New Haven, CT, USA, 3Nanobiology Institute, Yale University, West Haven, CT, USA, 4 Chemical Engineering, Columbia University, New York, NY, USA. Fusion between membrane-enveloped viruses and their hosts is a critical step in infection, enabling the virus to release its genome and subsequently hijack the host cell’s replication machinery. The genomes of enveloped viruses are packaged into multiple bulky ribonucleoprotein (RNP) particles whose release requires the dilation of the fusion pore - the initial, narrow, connection between fusing membranes. Electrophysiological studies of influenza A hemagglutinin (HA)-mediated fusion between HA-expressing fibroblasts and red blood cells and between HA-fibroblasts and voltage-clamped suspended bilayers comprise the most complete information to date about the dynamics of viral fusion pores. Small pores (~2-5nm) were found to flicker repetitively and/or fluctuate in size prior to terminal dilation. A quantitative understanding of the biophysical driving forces of viral membrane fusion, including membrane contact area, membrane tension and protein fusogen cooperativity, however, remains elusive, and a number of questions - regarding the physical basis of pore flickering and the number of HA trimers required for successful pore dilation - remain open. Here we describe a novel electrophysiological approach, which has been successfully applied to study SNARE-mediated fusion pores, to probe the HA-mediated fusion pore. We voltage-clamped HA-expressing fibroblasts in the cell-attached configuration and included nanodiscs (NDs) in the patch pipette as fusion partners to probe fusion pore nucleation and evolution at single pore resolution. NDs are flat, bilayer discs whose composition and size can be varied. Current through voltage-clamped fusion pores reports pore size with sub-millisecond time resolution. Preliminary data shows that isolated fusion pores flicker, are long-lived (~10s) and are of diameter ~1.5nm. 2046-Pos Board B183 Effect of Cholesterol Depletion on HA Distribution in the Viral Membrane of Influenza Rebecca A. Dunning, Marta K. Domanska, Kelly Dryden, Mark Yeager, Peter M. Kasson. Molecular Physiology and Biophysics, University of Virginia, Charlottesville, VA, USA. Influenza virus enters cells via a process of membrane fusion mediated by the hemagglutinin coat protein. We have previously shown that depletion of cholesterol from the viral envelope increases the rate of fusion despite decreasing fusion efficiency. Furthermore, this increased rate of fusion is only observed when cholesterol is removed from the viral envelope, not in the target membrane. Here we use cryo-electron microscopy to measure how the lateral distribution of hemagglutinin in influenza virions changes upon cholesterol extraction. Following removal of cholesterol from the viral envelope, hemagglutinin trimers move closer together. We estimate that HA trimer-trimer spacing decreases from 92.6 angstroms to 84.9 angstroms. Upon re-addition of cholesterol, hemagglutinin trimer spacing expands once Tuesday, February 10, 2014 again. Current models from single-virus microscopy suggest that fusion requires the engagement of several hemagglutinin trimers in close proximity. Our findings are in agreement with these models. If hemagglutinin trimers indeed shift closer together in the viral envelope upon cholesterol extraction, this would result in an increase in the rate of fusion. 2047-Pos Board B184 Cholesterol and Influenza Viral Fusion Mechanisms: Using Sterol Analogues to Probe Mechanism Katarzyna E. Zawada, Dominik Wrona, Marta K. Domanska, Peter M. Kasson. Department of Molecular Physiology and Biological Physics, University of Virginia, Charlottesville, VA, USA. Fusion between the viral envelope and target cell membrane is a crucial step for infection of influenza and other enveloped viruses. The influenza envelope protein hemagglutinin provides a necessary driving force for fusion. However, both viral and target membrane composition can also have a significant impact on infectivity. Here, we examine the effect of sterol composition on viral membrane fusion kinetics. We have previously shown that changes to cholesterol concentration in either synthetic target liposomes or viral membranes can alter both hemi-fusion and fusion kinetics. Increasing cholesterol in target liposomes caused a monotonic increase in fusion rate, while depleting viral cholesterol from the starting cholesterol:phospholipid ratio of 1:1 had a more complex effect. To further characterize what chemical properties of cholesterol are responsible for the observed changes and begin to dissect the responsible mechanism, we examined the effect of other related sterols. We tested seven cholesterol analogues, measuring fusion kinetics when each sterol was used in place of cholesterol in target liposomes or when cholesterol was extracted from the viral membrane and replaced by each sterol tested. As measured by fluorescence dequenching kinetics, fusion between X-31 influenza virus and target vesicles containing 20% sterol did not significantly change hemi-fusion or fusion rates among the sterols tested. Most sterols tested in the viral envelope had a minimal effect on fusion kinetics compared with cholesterol. However, replacing viral envelope cholesterol with cholesteryl sulfate significantly slowed both hemifusion and fusion. We are currently working to identify the chemical basis for this effect. 2048-Pos Board B185 Characterization of HIV-1 Entry Site Specificity using Single-Particle Tracking Chetan Sood, Mariana Marin, Gregory B. Melikian. Pediatric Infectious Diseases, Emory University, DECATUR, GA, USA. The first critical step in the HIV-1 infectious cycle is fusion of the viral membrane with the membrane of the host cell and penetration of the viral capsid into the host cytosol. Because the functionality of the HIV-1 fusion glycoprotein (Env) is pH-independent, it was thought that productive HIV-1 fusion occurs at the plasma membrane (PM). This hypothesis is supported by the observations that HIV-1 mediates fusion between adjacent cells and that cell-cell fusion occurs between Env and receptor/co-receptor expressing cells. To the contrary, it was recently demonstrated that, after engaging receptor/co-receptor at the cell surface, HIV-1 traffics via an endocytic pathway before fusing with the endosomal membrane. However, the previous virus labeling techniques were unable to reliably detect fusion with the PM. Here, we directly quantified the fraction of HIV-1 virions that fuse with the PM by co-labeling viral particles with a pHsensor incorporated into the viral membrane and a content marker that is released into the cytoplasm upon fusion. In imaging viruses bound to living cells, virus fusion at neutral pH is manifested as loss of the viral content marker without change to the signal from the pH-sensor. Upon virus entry to an acidic compartment, the reference signal from the pH-sensor is completely quenched, thus precluding detection of subsequent fusion. We found that only a small fraction of fusion events occur at neutral pH, presumably at the cell surface or in early, pH-neutral vesicles. Our finding implies that the majority of HIV-1 virions enters and fuses after trafficking into acidic compartments. Notably, the viral pH sensor also revealed occasional recycling of HIV-1 particles to the cell surface followed by their re-internalization. This work was supported by NIH R01 GM054787. 2049-Pos Board B186 Recognition of Lipid Domain Boundaries by the HIV Fusion Peptide is an Essential Step for HIV Membrane Fusion Sung-Tae Yang, Volker Kiessling, Lukas K. Tamm. University of Virginia, Charlottesville, VA, USA. ‘‘Lipid rafts’’ which are cholesterol-enriched regions of the plasma membrane have been recognized as possible platforms for HIV entry. However, due to their highly dynamic and nanoscopic nature in biological membranes, it is challenging 407a to investigate the role of lipid rafts in HIV-host interactions. In this study, we have used model systems with microscopic raft-like domains (Lo phase) in supported lipid bilayers and giant unilamellar vesicles mimicking HIV envelopes and T-cell membranes. We show that the phase separation of HIV or T-cell lipid mixtures is cholesterol-dependent, and membrane binding and lipid mixing are much more efficient in vesicles with coexisting Ld and Lo phases than those with single Ld or Lo phase, indicating that lipid phase separation is necessary and sufficient for efficient membrane fusion. Interestingly, time-resolved TIRF microscopy demonstrates that the HIV fusion peptide preferentially targets Lo/Ld boundary regions and promotes membrane fusion at the interface between Lo and Ld phases. Analysis of individual fusion events shows that pure Ld phase vesicles proceed to hemifusion and only vesicles with Lo/Ld phase boundaries fuse fully. Based on our designed minimal systems for understanding lipid raft-dependent HIV-host interactions, we propose that recognition of domain boundaries by the HIV fusion peptide is an essential step for HIV entry. 2050-Pos Board B187 Post Fusion Structure of the Transmembrane Domain of the Ebola Virus Surface Glycoprotein Jinwoo Lee, Lukas K. Tamm. University of Virginia, Charlottesville, VA, USA. Ebola virus (Ebov) is an enveloped virus causing hemorrhagic fever in humans and non-human primates with extremely high fatality rates. Fusion occurs when the virus reaches the endosomal compartment where the surface glycoprotein (GP) undergoes proteolytic cleavage and rearrangement. In this study we focus on the role of the transmembrane (TM) and membrane proximal (MPER) domains of Ebov GP in the fusion process. NMR spectroscopy was performed on a construct comprising Ebov TM and MPER (residues 632-676) to investigate its structure and role in fusion. Solution NMR studies show that the structure is composed of two helices. A dynamic N-terminal region (residues 632-642) is followed by a short MPER helix residing on the surface of the membrane (residues 643-651), a turn (residues 652-657), and the TM helix (residues 658-676). HSQC spectra of Ebov TM/MPER at pre- and post-fusion pH were similar suggesting a pH-independent role of this domain in fusion. Titration experiments revealed a binding site between the FL and TM domains at pH 5.5. The binding site is rich in aromatic residues on the TM (WTGW) and FL (YWTTQD) side. Liposome fusion assays showed that lipid mixing was enhanced when the liposomes contained Ebov TM/MPER. Taken together, we conclude that the Ebov TM/MPER and FL domains cooperate in fusion, but that only the FL structure [see refs. 1 and 2] responds to pH in this process. 1. Gregory et al. PNAS (2011) 108:11211-11216. 2. Gregory et al. J. Virol. (2014) 88:6636-6649. 2051-Pos Board B188 The Role of Acidic pH in Ebola Mediated Cell-Cell Fusion Ruben Markosyan1, Grigory Melikyan2, Shan-Lu Liu3, Fred Cohen4. 1 Rush University Medical Center, Chicago, IL, USA, 2Pediatrics, Division of Infectious Diseases, Emory University, Atlanta, GA, USA, 3Molecular Microbiology and Immunology, University of Missouri, Columbia, MO, MO, USA, 4Molecular Biophysics and Physiology, Rush University Medical Center, Chicago, IL, USA. The low pH within endosomes promotes fusion of many viruses, including Ebola virus (EBOV) from within this compartment. We have developed a system that monitors aqueous and lipid dye mixing to measure fusion between cells that is mediated by the fusion protein, GP, of EBOV. Fusion is pHdependent, and the extent of both aqueous and lipid dye mixing exhibits a maximum at pH 5.7. Some fusion occurs at neutral pH. Fusion requires cleavage of EBOV GP into two linked subunits. In a biological setting, cathepsins, whose activities are pH-dependent, are the responsible cleaving enzymes. Inhibiting cathepsins activity eliminated fusion. Introducing a recombinant cathepsin into the external solution largely restored pH-dependent fusion. In the laboratory, proper cleavage of EBOV GP can be achieved by using thermolysin and this treatment doubles the rate and extent of cell-cell fusion above that achieved by relying on endogenous cathepsins alone. For thermolysin-treated effector cells expressing EBOV GP, cathepsin inhibitors reduce the extent of fusion by roughly a factor of two. We have shown that EBOV GP mediated cell-cell fusion is reduced by proteinase K treatment, and have determined the stage of fusion at which EBOV GP becomes sensitive to this protease. We found that the EBOV GP fusion pore is initially small, and unlike other viral protein mediated fusion pores, the pore tends to remain small and typically does not enlarge. In addressing how acidic pH promotes EBOV GP mediated fusion, a mechanism which is somewhat controversial, we found that contributing equally are the increases in activity of cathepsins with lowered pH and the direct effects of low pH in promoting conformational changes of cleaved GP. Supported by NIH RO1 GM101539. 408a Tuesday, February 10, 2014 2052-Pos Board B189 Real-Time Imaging Reveals that HIV-1 Vpr Dissociates from the Core and Accumulates in the Nucleus after Viral Fusion Tanay M. Desai, Mariana Marin, Gregory B. Melikyan. Department of Pediatric Infectious Diseases, Emory University, Atlanta, GA, USA. Viral protein R (Vpr) is an HIV-1 accessory protein that associates with capsids during viral assembly and is important for infections in non-dividing cells. Vpr functions in host cells include induction of G2 cell-cycle arrest, and regulation of cellular proliferation and apoptosis. Vpr has two nuclear localization sequences that direct its transport to the nucleus. Fluorescently labeled Vpr (YFP-Vpr) is widely used to visualize HIV-1 cores in the cytoplasm during entry. Here we report on the dissociation of YFP-Vpr from HIV-1 cores shortly after viral fusion and its subsequent accumulation in the nuclei. Real-time live cell imaging showed that, under conditions of productive entry and infection, Vpr dissociated from cores post-fusion and accumulated in nuclei over time-scales that correlated with the kinetics of viral fusion (t1/2~15 min). Nuclear accumulation of Vpr scaled with the number of cell-bound virions and could be blocked by lysosomotropic agents or a fusion-inhibitory peptide. These effects were observed in two cell lines and were independent of the fusion proteins incorporated into viral particles. Fluorescence recovery after photobleaching of YFP-Vpr within the nucleus revealed quick (t1/2~3 min) recovery, indicating that Vpr dissociation from capsids is a rate-limiting step in Vpr post-fusion transport. Fluorescence correlation spectroscopy measurements on post-fusion nuclear YFP-Vpr, yielded fast and slow diffusive components (D~10 mm2/s and 0.8 mm2/s, respectively) similar to those measured for YFP-Vpr over-expressed in cells. These diffusion coefficients reflect that nuclear Vpr exists in two forms - as a monomer, and in large complexes with host proteins or perhaps even chromatin structures. Current efforts are underway to explore the determinants of the stability of Vpr-capsid complexes. This work was supported by the NIH R01 GM054787 grant. 2053-Pos Board B190 Delivery of Liposomal Contents to Outer Membrane Vesicles from Gram Negative Bacteria Michael Ficurilli, Carol Liu, Christopher Riviello, Maria Jose Pozo, Paul R. Meers. Rutgers University, New Brunswick, NJ, USA. Gram negative bacteria produce small ~50-200 nm vesicles from their outer membranes. These outer membrane vesicles (OMV) have been implicated in activities such as transmission of virulence factors, horizontal gene transfer and development of biofilms. In this investigation, we continue our studies on the association and/or fusion of various liposomes with OMV. The delivery of large encapsulated molecules into OMV from L. enzymogenes C3 was investigated using liposomes with lipid compositions previously observed to be apparently fusogenic (Bartos et al., Biophys. J. 104(2) suppl1, 90a). Liposomes (100 nm) composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(10 -racglycerol) (POPG) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) in a 1:3 ratio or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) were used to encapsulate dextran conjugates of Texas red averaging 40 kDa. They were incubated with Lysobacter OMV (30 C, 1 hr.), then sedimented through 15% iodixanol, and the fluorescence monitored as indicatiive of transfer of liposomal contents to fused products. Both liposomal compositions showed significant evidence of dextran transfer. Because biofilms also contain OMV, the interaction of these liposomes with E. coli (DH10B) biofilms was also investigated via fluorescence microscopy. Significant penetration and binding within the biofilm mass was observed, as well as possible fusion with OMV, and rarely, evidence of transfer of dextran into whole bacterial cells. Fluorescence resonance energy transfer (FRET)-based assays also demonstrated that liposomes as small as 30 nm could rapidly fuse with Lysobacter OMV, suggesting possible delivery to OMV with smaller perturbation and better biofilm penetration. 2054-Pos Board B191 Fusion Fore Dilation by Snare Proteins Zhenyong Wu1, Oscar Daniel Bello2, Sarah Marie Auclair2, Wensi Vennekate1, Shyam Sundar Krishnakumar2, Erdem Karatekin1. 1 Department of Cellular and Molecular Physiology, Yale University, New Haven, CT, USA, 2Department of Cell Biology, Yale University, New Haven, CT, USA. Hormones and neurotransmitters are released through exocytotic fusion pores (FPs) that can flicker open and shut multiple times, fluctuate in conductance, and either dilate or reseal irreversibly. FP properties determine the size and the amount of cargo released, and the time course of release, which modulate downstream effects. Fusion is driven by formation of a four-helix complex be- tween the neuronal/exocytotic vesicle-associated v-SNARE VAMP2/ synaptobrevin-2 (VAMP2) and its cognate plasma-membrane t-SNARE partner composed of syntaxin-1 and SNAP25. Pore nucleation requires zippering between the v- and t-SNAREs, but what molecular factors govern the subsequent pore dilation is not understood. Here, using conductance measurements across voltage-clamped single FPs we show that multiple SNARE proteins cooperate to dilate metastable FPs. We isolated flickering fusion pores in a biochemically defined assay where v-SNARE-reconstituted bilayer nanodiscs (vNDs) fuse with cells ectopically expressing cognate, ‘‘flipped’’ t-SNAREs. Using newly developed, large NDs that are 21-27 nm in diameter, we varied v-SNARE copy numbers from zero to up to 11 per ND face. Pore nucleation required a minimum of 2, and reached a maximum above ~4 copies per face, but the probability of pore dilation was far from saturating at 11 copies, the maximum that the NDs could hold per face. Our results indicate that copy numbers of available SNAREs may be pivotal in determining whether neurotransmitters or hormones are released through a transient (kiss & run) or an irreversibly dilating pore (full fusion) and provide a rationale as to why synaptic vesicles carrying 70 copies of v-SNAREs dock onto sites where as many t-SNAREs are clustered while only a few SNARE complexes are apparently enough to achieve fusion. 2055-Pos Board B192 Control of Fusion Pore Nucleation and Dynamics by SNARE Protein Transmembrane Domains Zhenyong Wu1, Sarah M. Auclair2, Oscar D. Bello2, Wensi Vennekate1, Shyam Krishnakumar2, Erdem Karatekin1. 1 Cellular and Molecular Physiology, Yale University, New Haven, CT, USA, 2 Cell Biology, Yale University, New Haven, CT, USA. Membrane fusion is a fundamental biological process, whose initial stages have been observed in hormone-secreting cells and neurons using electrophysiological and electrochemical methods. The initial connection between the plasma membrane and a hormone- or neurotransmitter-filled vesicle -the fusion porecan flicker open and closed repeatedly before dilating or resealing irreversibly. Pore dynamics determine events such as vesicle recycling and release kinetics, but pore properties are poorly known, because fusion pores are transient, and biochemically defined assays with single-pore sensitivity are lacking. We isolated single flickering pores connecting v-SNARE-reconstituted nanodiscs to cells ectopically expressing cognate, ‘‘flipped’’ t-SNAREs, voltage-clamped in the cell-attached configuration. Currents through such pores directly report sub-millisecond single-pore dynamics. We found that interactions between vand t-SNARE transmembrane domains (TMDs) observed in a recent crystal structure promote, but are not essential for pore nucleation. Surprisingly, TMD interactions also affected pore lifetimes. Rod-shaped, post-fusion cisSNARE complexes vacated the highly curved fusion site where they fit poorly, leaving pore properties to be determined largely by lipid bilayers. In contrast, Y-shaped mutants deficient in TMD-zippering lingered at the fusion site, preventing pore resealing for >60 s. Thus, post-fusion geometry of the proteins determines pore stability, analogous to the well-known effects of lipid geometry on highly curved fusion intermediates. 2056-Pos Board B193 Snare Mediated Fusion with Membrane Tension Control Joerg Nikolaus, Erdem Karatekin. Cellular and Molecular Physiology, Yale School of Medicine, New Haven, CT, USA. Fusion of membranes is ubiquitous in life. It is essential for neurotransmitter and hormone release, intracellular vesicular trafficking, fertilization, and viral infection. SNARE proteins constitute a highly conserved minimal fusion machinery mediating intracellular membrane merger from slow fusion of large yeast vacuoles (minutes) to extremely fast neurotransmitter release (<1 ms). While membrane tension was suggested to inhibit fusion by suppressing dimpling of membranes by viral fusion proteins (Markosyan et al., Biophys J, 1999), it was suggested to promote fusion pore opening and dilation in other studies (Shillcock and Lipowsky, Nat Mater, 2005; Nikolaus et al., Biophys J, 2010; Warner and O’shaughnessy, Biophys J, 2012). Thus, membrane tension may affect distinct stages of the fusion process differentially. To resolve how tension affects fusion, we established a fusion assay in which membrane tension is precisely controlled. Our approach is based on a previously established bulk assay in which v-SNARE reconstituted small liposomes (vSUVs) fuse to tSNARE containing giant unilamellar vesicles (tGUVs, ~10-30 micrometers in diameter) (Malsam et al., EMBO J, 2012). Using a micropipette, a single GUV is picked up, whose membrane tension is controlled by the aspiration pressure. Another pipette is maneuvered nearby to puff a suspension of vSUVs. Fusion is monitored as an increase of the GUV tongue projection in the aspiration pipette whose position can be determined with sub-pixel resolution. Because the Tuesday, February 10, 2014 micromanipulation setup is mounted on a spinning disc confocal microscope, simultaneous monitoring of fluorescence from labeled membranes can also be used to probe vesicle docking and fusion. Our preliminary results show increasing membrane tension increases the fusion rate. 2057-Pos Board B194 Collective Action of SNAREpins Exerts Forces between Membranes that Activate Fusion Hakhamanesh Mostafavi1, Ben Stratton1, Jason M. Warner1, Erdem Karatekin2, Ben O’shaughnessy1. 1 Chemical Engineering, Columbia University, New York, NY, USA, 2 Cellular and Molecular Physiology, Yale University, New York, NY, USA. SNARE proteins mediate most intracellular membrane fusion processes such as exocytosis. It has been established that vesicle associated v-SNAREs and target-membrane associated t-SNAREs, assemble into a parallel four-helix bundle (SNAREpin) in a zipper like fashion, which brings the membranes close together. However, the detailed mechanisms whereby SNAREs drive fusion remains unclear. It is also unknown whether several SNAREpins cooperate to induce fusion, and widely varying SNARE requirements for fusion are reported, 2-15 in vivo (Montecucco et al., Trends Biochem Sci, 2005) and 1-11 in vitro (van den Bogaart et al., NSMB, 2010; Karatekin et al., PNAS, 2010). Here, we developed a mathematical model of SNAREpins connecting a vesicle to a planar membrane, quantifying inter-membrane, inter-SNAREpin and membrane-SNAREpin interactions, and taking account of the zippering/ unzipping of the SNAREpins. Monte Carlo simulations showed that SNAREpins assemble through the SNARE motifs and self-organize into a ring. The ring tends to expand, driven by inter-SNAREpin and SNAREpin-membrane interactions, reducing membrane separation by geometrical coupling. Assuming an energy criterion for fusion, we determined the waiting times for fusion from the distributions of inter-membrane energies. Our data show that although one SNAREpin can induce membrane fusion, fusion waiting times decrease rapidly with the number of SNAREpins. Applying the model to single-vesicle fusion assays, we predict that the dependency of docking-to-fusion delay times on the number of v-SNAREs reaches a plateau, in agreement with experiments (Karatekin et al., PNAS, 2010). We also find that waiting times increase with vesicle size and with insertion of flexible segments into the SNARE linker domains, in qualitative agreement with experiments (McNew et al., Mol Cell, 1999). Our results suggest that fully zippered SNAREs work in concert to trigger fusion, and explain the wide range of reported SNARE requirements for fusion. 2058-Pos Board B195 Cholesterol Modulates SNARE Mediated Hemi- and Full-Fusion Alex J.B. Kreutzberger, Volker Kiessling, Lukas K. Tamm. University of Virginia, Charlottesville, VA, USA. Cholesterol is essential for exocytosis in secretory cells, but distinguishing contributions from lateral organization and dynamics of membrane proteins and stabilization of fusion pores by intrinsic curvature and other mechanical effects of cholesterol have been elusive. The direct effect of cholesterol on fusion pore formation was examined between synaptobrevin 2 (VAMP 2) containing proteoliposomes and an acceptor SNARE complex containing planar supported bilayer using both membrane and content fluorescent markers. This revealed that increasing cholesterol in either the planar supported bilayer or in the synaptobrevin proteoliposome decreases the amount of hemi-fusion and increases the amount of full-fusion with minimal effects on the fusion kinetics. 2059-Pos Board B196 Chasing the Functional Asymmetry between C2A and C2B in Full-Length Synaptotagmin 1 during Ca2D-Dependent Membrane Binding Volker Kiessling1, Bin Lu2, Lukas K. Tamm1, David S. Cafiso2. 1 Molecular Physiology and Biological Physics, University of Virginia, Charlottesville, VA, USA, 2Chemistry, University of Virginia, Charlottesville, VA, USA. Synaptotagmin 1 (Syt1) acts as the major calcium sensor in neuronal exocytosis. Syt1 is a synaptic vesicle-anchored membrane protein that contains two tandem Ca2þ binding C2 domains, named C2A and C2B. There is evidence that Syt1 interacts with membrane lipids and SNARE proteins simultaneously to facilitate two key steps during Ca2þ-triggered membrane fusion: vesicle binding and content release. Although the overall function of Syt1 has been extensively investigated, the detailed molecular behavior of the single C2A and C2B domains in the neural regulatory process remains unclear. In particular, the differential function of the two C2 domains and how they cooperate in membrane fusion has not been determined. 409a Here we employed EPR spectroscopy, fluorescence interference contrast (FLIC) microscopy and total internal reflection fluorescence (TIRF) microscopy to dissect the state of C2A and C2B domains in full-length Syt1 during Ca2þ-dependent membrane binding. CW-EPR lineshapes and power saturation of spin-labeled positions in calcium binding loops of C2A in full-length Syt1 (1-421) and truncated Syt1 (1-266) without C2B domain suggest that C2A domain alone in Syt1 has a stronger membrane binding ability. A TIRF liposome capture assay further reveals that truncated Syt1 has a significant higher initial rate and extent in trans-binding with liposomes than full-length Syt1. Our data indicate that the C2B domain might be involved in some cis-binding thereby reducing total liposome binding. FLIC microscopy validates the strong involvement of C2A during Ca2þ-dependent liposome binding. Our detailed structural and functional information thus provides a clue to differential regulatory mechanisms employed by C2A and C2B domains in fulllength Syt1 interacting with Ca2þ and key membrane lipids, such as PS and PIP2. 2060-Pos Board B197 Mechanical Model for Self-Assembly of Synaptotagmin on a Lipid Membrane Jie Zhu, James E. Rothman. Cell Biology, Yale University, New Haven, CT, USA. Synaptotagmin (Syt) is a calcium-sensor that is responsible for the actionpotential-controlled fusion of synaptic vesicles to pre-synaptic membranes. Recent biochemical and structural studies show that Syt can form ring-like oligomers, which occasionally convert into tubular structures on monolayer or bilayer membrane with buckled membrane inside. This suggests certain mechanical interactions between the Syt and the lipid bilayer. To explore it in detail, we developed a coarse-grained mechanical model assuming that (i) Syt self-polymerizes into an elastic chain with a spontaneous curvature; (ii) Syt attracts the membrane through binding sites located at the inner side of the Syt molecule; and (iii) membrane is a uniform sheet with constant bending rigidity and tension. Using computer simulations, we have been able to estimate the spontaneous curvature and bending stiffness of the Syt chain. The model also allowed us to understand how the Syt oligomerization depends on the strength of Syt-membrane adhesion, the bending rigidity of the membrane, and the pressure on the membrane. These experimentally testable predictions from this modelling study will provide insight into the molecular mechanism of calcium-triggered membrane fusion at the synapse. 2061-Pos Board B198 Single Vesicle Assay to Study Membrane Tethering and Docking Factors Jiajie Diao. Stanford and HHMI, Stanford, CA, USA. In vitro vesicle-vesicle fusion assays based on fluorescence resonance energy transfer (FRET) of lipid or content mixing are widely used to investigate the molecular mechanism of membrane fusion process. However, without FRET signals from lipid or content mixing, these ensemble assays are insensitive to early stages of fusion such as tethering and docking. We have developed a single vesicle assay to study protein factors involved in tethering and docking [1-3]. Through our assay, we have studied proteins inducing membrane aggregation [4] and enhancing SNARE-mediated vesicle docking [5-6]. 1. Diao, J. et al. Nat. Protoc. 7, 921 (2012). 2. Diao, J. et al. Bioessays 35, 658 (2013). 3. Kyoung, M. et al. Nat. Protoc. 8, 1 (2013). 4. Diao, J. et al. Langmuir 25, 7177 (2009). 5. Diao, J. et al. J. Am. Chem. Soc. 135, 15274 (2013). 6. Lai, L. et al. Elife 3, e03756 (2014). 2062-Pos Board B199 Deficiency of HID-1 Leads to Impaired Proinsulin Processing Wen Du, Tao Xu. Chinese Academy of Sciences, Institute of Biophysics, Beijing, China. Diabetes mellitus has been a major social problem due to its prevalence, which results from a combination of peripheral insulin resistance and insufficient insulin secretion.The whole process of insulin biosynthesis, transportation, maturation and secretion is largely understood, however, detailed mechanism remains to be uncovered. We reported here a highly conserved protein, HID1, functions during the correct processing of insulin. We generated a beta cell-specific conditional knock out mouse model of hid-1 gene. The knock out mice showed significant glucose intolerance while normal response to insulin with no significant defect in islets’ morphology. Further study revealed remarkable increase in proinsulin to insulin ratioand abnormal proinsulin accumulation, suggesting that HID-1 may function in the conversion process from proinsulin to insulin. 410a Tuesday, February 10, 2015 Membrane Structure II 2063-Pos Board B200 Role of Headgroup Dipole Interactions in Phosphatidylcholine and Phosphatidylserine Bilayers Hongcheng Xu1, Sai Ganesan2, Silvina Matysiak1. 1 Fischell Department of Bioengineering and Institute for Physical Science and Technology, University of Maryland, College Park, MD, USA, 2Fischell Department of Bioengineering, University of Maryland, College Park, MD, USA. The presence of a dielectric gradient in lipid bilayers is critical for membrane peptide folding in vivo. In addition, the hydrogen bonding ability of lipid head groups is known to affect bilayer properties. To reveal the role of headgroup dielectric properties in phospholipid bilayers, we have developed a polarizable Coarse-Grained (pCG) model, where the dipole moment of polar groups, such as serine and ester groups, can be adjusted through the addition of two extra ‘‘dummy’’ particles inside a CG bead. These dummy particles can mimic the hydrogen bonding network that can exist among head groups. The addition of polarization effects into CG beads has been proven effective in prior work regarding de novo peptide secondary structure folding with no external bias (Ganesan, 2014). In this work, we have explored the role of dipole interactions in monocomponent lipid bilayers using POPC (Palmitoyl Oleoyl Phosphatidyl-Choline) and POPS (Palmitoyl Oleoyl Phosphatidyl-Serine) lipids. An excellent agreement is observed on several structural and dynamical bilayer properties when compared to all-atom simulation data. We will present results on the role of dipole-dipole and dipole-charge interactions in shaping the clustering of PS lipids. Since dipole interactions are influenced by charge screening, the model is sensitive to changes in salt concentration. Our results suggest that headgroup dipole interactions may participate in PS/PC lipid phase separation as observed in experiments. Reference ‘‘The role of backbone dipole interactions in the formation of secondary and super-secondary structures of proteins’’, Ganesan S. J., Matysiak S., Journal of Chemical Theory and Computation (2014). 2064-Pos Board B201 Domain Formation in Quarternary Lipid Bilayer System: A CoarseGrained Molecular Dynamics Study Shushan He, Lutz Maibaum. Department of Chemistry, University of Washington, Seattle, WA, USA. The spatial organization of phospholipids and sterols in cellular membranes is believed to play an important role in membrane function and cell activities. Both recent experimental and simulation studies have suggested various possible heterogeneous spatial distributions of lipids and cholesterol, including the formation of nanoscale lipid domains, in multi-component bilayer systems. In this study, we use coarse-grained Molecular Dynamics simulations to study the phase behavior of quarternary lipid mixtures as a function of membrane composition. We focus on the formation and size of nanoscopic lipid domains, their compositions, and their signatures in observables such as partial density correlation functions and structure factors. 2065-Pos Board B202 Investigating Lipid Phase Changes from Liquid Crystalline to Ripple to Gel Phases with All-Atom Molecular Dynamics Simulations Pouyan Khakbaz1, Jeffery Klauda2. 1 University of maryland college park, College Park, MD, USA, 2Chemical and Biomolecular engineering, University of maryland college park, College Park, MD, USA. Lipid mixed with water can exist in many different phases that depends on various factors, such as hydration, temperature and pressure (J. Chem. Theory Comput.,6 (8): 2488 (2010)). The liquid crystalline (chain-disordered state) is a well-studied phase for single lipid bilayers both experimentally and computationally. At low enough temperatures or hydration levels, this chain-disordered state can change to a gel state with high chain order and a certain chain tilt with respect to the membrane normal. Depending on the lipid, an intermediate phase can exist between the gel and liquid crystalline phase, which is known as the ripple phase. The main objective of this study was probing the accuracy of the CHARMM36 (C36) force field (FF) (J. Phys. Chem. B.,114 (23): 7830 (2010)) in predicting phase changes in lipid bilayers. Pure and mixed lipid bilayers at different compositions of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and dipalmitoylphosphatidylcholine (DPPC) were studied using molecular dynamics (MD) simulations. Based on the pure gel-phase transition temperature of each lipid, a range of temperatures was selected (from 200C to 400C) (Biochem. 18: 3280 (1979)). MD simulations were able to capture ripple phase in %25 DMPC and %75 DPPC mixture and pure DMPC at 250C. Simulations were run for 200-300 ns. MD simulations also show either a gel state or gel-like state depending on if the system gets trapped (gel-like is a state without proper leaflet alignment). Phase diagram was compared to that obtained from experimental data such as NMR. Based on the phase diagram and our transition temperatures, C36 FF accurately predicts the phase transitions of this fully saturated PC lipids. Therefore, studies on phase coexistence of liquid ordered and liquid disordered domains are likely to be accurate using the C36 FF. 2066-Pos Board B203 Molecular Dynamics Simulations of Sphingomyelin-Cholesterol Bilayers Hojin Kang, Jeffery B. Klauda. Chemical and Biomolecular Engineering, University of Maryland College Park, College Park, MD, USA. Sphingolipids are one of the major components in animal cell membranes. One type of sphingolipids is sphingomyelins which are abundant in myelin sheath surrounding nerve cell axons. The two common forms of sphingomyelin targeted in this research are palmitoylsphingomyelin (PSM) and stearoylsphingomyelin (SSM). Another bilayer component included in our membranes that has high affinity to sphingomyelins is cholesterol, which aids in maintaining membrane fluidity and structural integrity. Sphingomyelins and cholesterol make up a great part of highly dynamic membrane domains, termed lipid rafts. With that being said, this work tests the accuracy of molecular dynamics (MD) simulations with the CHARMM36 (C36) lipid force field for PSM and SSM, each SM bilayer has varying concentrations of cholesterol. Properties such as surface area per lipid, x-ray and neutron form factors, and chain deuterium order parameters are to be compared to those yielded from past experiments to prove C36’s ability to correctly simulate the mixed SM-cholesterol lipid bilayers. Based on past studies on C36 lipid force field, we expect to see excellent agreement between experimental and MD simulation data for more complex mixtures of membranes that contain SM lipids. 2067-Pos Board B204 Influence of Cholesterol on Phospholipid Bilayer Dynamics Christopher T. Boughter, Jeffery B. Klauda. Chemical and Biomolecular Engineering, University of Maryland, College Park, MD, USA. Surrounding every living cell is a membrane comprised of an assortment of lipids, cholesterol, and proteins. Bilayers are dynamic systems with phospholipid and cholesterol molecules diffusing within a leaflet and occasionally flipping between leaflets. In biological systems, bilayers can exist in two phases; liquid ordered or liquid disordered. Molecular dynamics (MD) simulations give windows of insight into the biophysical properties of these bilayers. In this study, the effects of cholesterol on lipid bilayers are investigated by examining a variety of phospholipid head groups, cholesterol concentrations, chain saturations, and chain lengths. Two fatty acid chains, dimyristoyl (DM) and dioleoyl (DO), were used in our initial simulations with a phosphatidylcholine head group. The DM chain was then studied with several other phospholipid head groups including phosphatidic acid, phosphatidylethanolamine, phosphatidylglycerol, phosphatidylinositol and phosphatidylserine. In order to ensure that the MD simulations are physically accurate, the systems were checked at different simulation sizes as well as compared to earlier simulations, X-ray diffraction experiments, and NMR data. Simulations between 50 and 150ns at temperatures of either 303.15 or 333.15K elucidated the lipid ordering and phase behavior of these lipids in liquid disordered and liquid ordered phases. Increases in cholesterol concentration were seen to increase order in liquid disordered bilayers while simultaneously promoting disorder in bilayers which form gels in pure bilayers at temperatures of 303.15K. 2068-Pos Board B205 Microscopic Model and Analytic Derivation of Area Per Molecule for DPPC-Cholesterol Bilayers Boris B. Kheyfets, Sergei I. Mukhin. Theoretical Physics and Quantum Technologies, Moscow Institute for Steel and Alloys, Moscow, Russian Federation. An area per molecule dependence on components concentration for DPPCCholesterol bilayers is calculated analytically using a microscopic model in a biologically relevant concentration range. DPPC lipid is modeled as flexible string with finite bending rigidity [1,2]. Cholesterol molecule is modeled as rigid rod of finite thickness [3]. Surface tension at hydrophobic interface is linear combination of ‘‘partial tensions’’ of bilayer components. The model’s three important parameters are: surface tension at hydrophobic interface for pure DPPC membrane, bending rigidity of DPPC lipid, extrapolated surface tension at hydrophobic interface for cholesterol membrane. These parameters are chosen by nearly perfect fitting agreement of our theoretical curve with molecular dynamics simulations data [4] for these two-component bilayers. The molecular Tuesday, February 10, 2015 chains order parameter dependence on coordinate for 40% cholesterol - 60% DPPC membrane, measurable in NMR experiments, is calculated analytically and compared with molecular dynamics simulation data [5]. The order parameter calculation allows for DPPC tilt angle. The model parameters found by fitting the MD data are used further to calculate lateral pressure distribution and coefficient of thermal area expansion. The microscopic model allows one to study other thermodynamic coefficients and diffusion phenomena in multi-component bilayers. 1.Mukhin S.I., S. Baoukina, Analytical derivation of thermodynamic characteristics of lipid bilayer from a flexible string model. Phys. Rev. E. 71: 061918 (2005). 2.Mukhin S.I., B.B. Kheyfets, Analytical approach to thermodynamics of bolalipid membranes. Phys. Rev. E. 82: 051901 (2010). 3.Mukhin S.I., B.B. Kheyfets, Pore formation phase diagrams for lipid membranes. JETP Lett. 99: 358-362 (2014). 4. Edholm O., J.F. Nagle. Areas of Molecules in Membranes Consisting of Mixtures. Biophys. J. 89: 1827-1832 (2005). 5. Hofsass C., Lindahl E., and Edholm O. Molecular Dynamics Simulations of Phospholipid Bilayers with Cholesterol. Biophys. J. 84: 2192-2206 (2003). 2069-Pos Board B206 Simulation Study of Composition Fluctuations in Lipid Bilayers Svetlana Baoukina, Dmitri Rozmanov, D. Peter Tieleman. University of Calgary, Calgary, AB, Canada. Lipid bilayers constitute the basis of biological membranes. Understanding lipid mixing and phase behavior can provide important insights into membrane lateral organization (the raft hypothesis). Here we investigate model lipid bilayers below and above the miscibility transition temperatures. Molecular dynamics simulations with the MARTINI coarse-grained force field are employed to model bilayers on a length scale approaching 100 nm laterally and a time scale of tens of microseconds. We simulate lipid mixtures containing saturated and unsaturated lipids, and cholesterol at different concentrations and temperatures between 270 and 340 K. The coexistence of liquid-crystalline and gel, as well as liquid-ordered and liquid-disordered phases is reproduced. We induce a gradual transition from phase separation to mixing by raising the temperature and adding hybrid lipids (with a saturated and an unsaturated chains). The evolution of bilayer properties along this transition is analyzed. Domain size and phase boundary length, the length and time scales of composition fluctuations, and inter-leaflet coupling are quantified. The results allow characterizing partitioning of hybrid lipids between the coexisting phases, their role in composition fluctuations, and also the effect of spontaneous curvature on composition fluctuations. Curved domains are observed in both symmetric and asymmetric bilayers (with different composition of the two leaflets). 411a Tampere, Finland, 4Department of Chemical Engineering, Kyoto University, Kyoto, Japan, 5Fachbereich Physik, Freie Universita¨t Berlin, Berlin, Germany, 6IBCP, CNRS UMR 5086, Lyon, France, 7Aalto University, Espoo, Finland, 8Max Planck Institute for Chemical Energy Conversion, Mu¨lheim an der Ruhr, Germany, 9INSERM, U1134, DSIMB; Institut National de la Transfusion Sanguine (INTS); Laboratoire d’Excellence GR-Ex, Paris, France. We compare the C-H order parameters measured by Nuclear Magnetic Resonance (NMR) experiments to those predicted by 12 different molecular dynamics (MD) simulation models. We focus on the order parameters of the lipid headgroups and glycerol backbones in phospholipid bilayers. Only two of the models (CHARMM36 [1] and Maciejewski-Rog [2]) give a reasonable agreement with experiments for a fully hydrated lipid bilayer. We then compare (for the two best-performing models at full hydration and for the Berger model [3], the most used lipid model in the literature) to NMR experiments the changes in the order parameters as a function of hydration level, NaCl and CaCl2 concentrations, and cholesterol content. The results clearly show that the glycerol and headgroup structures in the Berger model are not realistic, the Na ion partitioning is significantly too strong and cholesterol-induced structural changes are overestimated. The CHARMM36 and Maciejewski-Rog perform better, but the Na partitioning is too strong at least in the latter. This is an open science project that is progressed at nmrlipids.blogspot.fi. All the results and discussions are available at that address. [1] J. B. Klauda, ..., R. W. Pastor. J. Phys. Chem. B 114 7830 (2010) [2] A. Maciejewski, M. Pasenkiewicz-Gierula, O. Cramariuc, I. Vattulainen, T. Rog. J. Phys. Chem. B 118 4571 (2014) [3] O. Berger, O. Edholm, F. Ja¨hnig. Biophys. J. 72 2002 (1997) 2070-Pos Board B207 Fatty Acid Interactions with RNA Building Blocks: Origin of Life Implications Parisa Akhshi, Peter Tieleman. Centre for Molecular Simulation and Department of Biological Sciences, University of Calgary, Calgary, AB, Canada. Several experimental and computational methods have been used to address important questions regarding fatty acid interactions with RNA building blocks that have implications in the origins of life1,2,3. A recent study by Keller et al.2 showed that nucleobases can bind to and stabilize the aggregation of prebiotic amphiphiles, which could support a possible mechanism for the emergence of protocells. Some nucleobases were found to bind stronger to the aggregates of putative prebiotic amphiphiles. Among carbohydrates, ribose has shown a greater potential to permeate through bilayers compared to its diastereomers. This is fundamentally interesting; as ribose/deoxy ribose are sugars found in RNA and DNA. The mechanisms, however, are not fully understood. We use molecular dynamics simulation to systematically study the permeation of furanose and pyranose carbohydrates as well as RNA nucleobases through different fatty acid bilayers as models of prebiotic conditions. The membrane fluidity and hydrogen bonding interactions have found to play significant roles in selective permeability. References: 1. Pohorille et al., ASTROBIOLOGY, 13(2), 177-188, 2013 2. Keller et al., PNAS, 110(33), 13272-13276, 2013 3. Szostak et al., PNAS, 102(17), 6004-6008, 2005 2072-Pos Board B209 Solid-State 2H NMR Investigation of Transducin Activation by Rhodopsin Xiaolin Xu1, Andrey V. Struts2,3, Aswini Kumar Giri2, Trivikram R. Molugu2, Charitha Guruge4, Samira Faylough4, Carolina L. Nascimento4, Nasri Nesnas4, Victor J. Hruby2, Michael F. Brown1,2. 1 Department of Physics, University of Arizona, Tucson, AZ, USA, 2 Department of Chemistry and Biochemistry, University of Arizona, Tucson, AZ, USA, 3Laboratory of Biomolecular NMR, St. Petersburg State University, St. Petersburg, Russian Federation, 4Department of Chemistry, Florida Institute of Technology, Melbourne, FL, USA. X-ray structures of active-state rhodopsin (Meta-II) and the cognate G-protein transducin are available, yet the transducin activation mechanism by rhodopsin is still obscure due to lack of atomistic dynamical information. We are studying the conformations of retinal in active Meta-II, and how the presence of the alltrans retinal agonist yields substantial differences in activation of transducin compared to opsin. Solid-state NMR spectroscopy gives information pertaining both to structures and dynamics, and is a powerful method to study how rhodopsin activates transducin. Experiments are currently underway with selectively deuterated retinal in aligned membrane samples containing active MetaII [1]. Simulation of the 2H NMR lineshape of the aligned samples in terms of a static uniaxial distribution reveals the bond orientations of retinal methyl groups and mosaic spread, which represents the alignment disorder of the stacked membranes [2,3]. Comparison with the solid-state 2H NMR spectra predicated by published X-ray results enables proposed structures for active Meta-II to be tested [4]. Moreover, the solid-state 2H NMR spectral lineshapes show the role of dynamical fluctuations of the protein. We are also conducting solid-state 2H NMR experiments with deuterated C-terminal peptide of transducin to study the interaction between the G-protein and the rhodopsin transmembrane helices. Our hypothesis is that association and dissociation cycles of transducin depend on the relationship between the local dynamics of peptide and the fluctuations of rhodopsin helices. Solid state 2H NMR experiments can not only tell us how rhodopsin activates transducin, but also can reveal the general mechanisms whereby GPCRs activate the cognate G-proteins. [1] A.V. Struts et al. (2011) PNAS 108, 8263. [2] X. Xu et al. (2014) Encycl. Mag. Res. 3, 275-286. [3] B. Mertz et al. (2012) BBA 1818, 241-251. [4] A.V. Struts et al. (2007) JMB 372, 50-66. 2071-Pos Board B208 Open Collaboration that uses NMR Data to Judge the Correctness of Phospholipid Glycerol and Head Group Structures in Molecular Dynamics Simulations Patrick F.J. Fuchs1,2, Matti Javanainen3, Antti Lamberg4, Markus S. Miettinen5, Luca Monticelli6, Jukka Ma¨a¨tta¨7, O.H. Samuli Ollila7, Marius Retegan8, Hubert Santuz2,9. 1 Institut Jacques Monod, CNRS, Paris, France, 2Universite´ Paris Diderot, Sorbonne Paris Cite´, Paris, France, 3Tampere University of Technology, 2073-Pos Board B210 n-3 PUFA-Containing Phospholipids Studied by MD Simulations: A Comparison of EPA, DPA and DHA Xiaoling Leng1, Jacob J. Kinnun1, Saame Shaikh2, Stephen Wassall1, Scott Feller3. 1 IUPUI, Indianapolis, IN, USA, 2Est Carolina University, Creenville, NC, USA, 3Wabash College, Crawfordsville, IN, USA. A wide range of health benefits is associated with consuming omega-3 polyunsaturated fatty acids (n-3 PUFA) from marine oils. Eicosapentaenoic acid 412a Tuesday, February 10, 2015 (EPA, 20:5), its metabolite docosapentaenoic acid (DPA, 22:5) and docosahexaenoic acid (DHA, 22:6) are the major n-3 PUFAs derived primarily from dietary consumption. Whether their effects on health are shared or complementary is a matter of current debate. To understand how molecular organization within cell membranes responds to the incorporation of each n-3 PUFA into phospholipids, we are performing MD simulations on model membrane systems composed of 1-stearoyl-2-eicosapentaenoylphosphatidylcholine (18:0-20:5PC), 1-stearoyl-2-docosapentaenoylphosphatidylcholine (18: 0-22:5PC), 1-stearoyl-2-docoshexaenoylphosphatidylcholine (18:0-22:6PC) and, as a monounsaturated comparison, 1-stearoyl-2-oleoylphosphatidylcholine (SOPC, 18:0-18:1PC) in the absence and presence of cholesterol. They are run at 37 C under constant pressure for 200 ns on bilayers containing 98 lipid molecules (single component membranes) or 100 lipid molecules (80 phospholipids and 20 cholesterols) that are hydrated in the ratio of 20 water molecules per lipid molecule. As validation, order parameter profiles along the sn-1 chain calculated from the trajectories will be compared with results from solid state 2H NMR experiments. Our analysis will identify the difference in flexibility between EPA, DPA and DHA chains. The impact of the difference in flexibility of each n-3 PUFA on the conformation of the adjacent stearic acid chain at the sn-1 position and on the interaction with cholesterol will then be investigated. 2074-Pos Board B211 DHA Disorders Raft-Like Domains as Revealed by Solid State 2H NMR Jacob J. Kinnun1, Justin A. Williams1, William Stillwell2, Robert Bittman3, Saame R. Shaikh4, Stephen R. Wassall1. 1 Department of Physics, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA, 2Department of Biology, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA, 3Department of Chemistry and Biochemistry, Queens College of CUNY, Flushing, NY, USA, 4 Department of Biochemistry & Molecular Biology and East Carolina Diabetes and Obesity Institute, East Carolina University, Greenville, NC, USA. Dietary omega-3 polyunsaturated fatty acids (n-3 PUFAs), such as docosahexaenoic acid (DHA, 22:6), have a wide variety of health benefits. However, the origin of the benefits at the molecular level is yet to be elucidated. A membrane-mediated mechanism in which n-3 PUFAs are incorporated into phospholipids and modulate molecular organization is one possibility. Cellular membranes are inhomogeneous where structurally diverse lipids can exist in separate domains. Regions rich in sphingomyelin (SM) and cholesterol, commonly called lipid rafts, contain important signaling proteins. In a recent solid-state 2H nuclear magnetic resonance (2H NMR) study of a model membrane composed of 1-[2H31] palmitoyl-2-docosahexaenoylphosphatidylcholine (PDPC-d31), a deuterated analog of a DHA-containing phospholipid, in mixtures with SM and cholesterol, we discovered that DHA could significantly enter raft-like domains. How DHA affects the molecular organization within the raft-like domains is addressed here by observing PSM-d31, an analog of SM with a perdeuterated N-palmitoyl chain. The 2H NMR spectra for PSM-d31 in mixtures with PDPC and cholesterol exhibit two spectral components, a larger more ordered component that we attribute to raft-like domains and a smaller less ordered component that we attribute to non-raft-like domains. On average, the order of PSM-d31 is reduced and, thus, disordering of PSM-d31 by PDPC is indicated. Our observations confirm that DHA can infiltrate rafts and affect molecular organization, which has implications for the signaling of raft and non-raft proteins. Furthermore, these results are consistent with in vivo studies showing that DHA infiltrates rafts. 2075-Pos Board B212 Disorderly Polyunsaturated Fatty Acids and Orderly Cholesterol: Just How do they get along in a Membrane? Denise V. Greathouse1, Jacob J. Kinnun2, Justin A. Williams2, Drew Marquardt3, Jeffrey B. Klauda4, Roger E. Koeppe II1, John Katsaras5,6, Thad A. Harroun3, Stephen R. Wassall2. 1 Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville, AR, USA, 2Department of Physics, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA, 3Department of Physics, Brock University, St. Catharines, ON, Canada, 4Department of Chemical and Biomolecular Engineering, University of Maryland, College Park, MD, USA, 5 Oak Ridge National Laboratory, Oak Ridge, TN, USA, 6Department of Physics, University of Tennessee, Knoxville, TN, USA. Polyunsaturated fatty acids (PUFA) are found in great abundance in neural membranes where they are essential for function. Their incorporation into membrane phospholipids has also been proposed to be responsible, in part, for a plethora of health benefits associated with dietary consumption of fish oils. Extremely high disorder that deters close proximity to cholesterol is what distinguishes PUFA from more common monounsaturated and saturated fatty acids. The aversion to cholesterol is exemplified by a solubility of only ~15 mol% in di-polyunsaturated 1,2-diarachidonylphosphatidylcholine (DAPC), as opposed to most phospholipid bilayers that can solubilize >50 mol%. According to our earlier neutron scattering experiments using deuterated analogs of cholesterol, the head group and tail of the sterol reside within the DAPC bilayer at the center. An orientation parallel to the plane of the bilayer and between leaflets is implied that, remarkably, differs from the generally accepted one in which the head group sits near the aqueous interface while the tail extends towards the middle of the bilayer. Here we present solid state 2H NMR spectra acquired with aligned multilayers that establish the bilayer normal constitutes the axis of motional averaging for a deuterated analog of cholesterol incorporated into DAPC. The dilemma that this result creates is being evaluated, including with the aid of molecular dynamics simulations. 2076-Pos Board B213 An Investigation of Whether Vitamin E Preferentially Interacts with Polyunsaturated Lipids Andres T. Cavazos1, Jacob J. Kinnun1, Justin A. Williams1, Bruce D. Ray1, Morris Bank1, Paul E. Harper2, Jeffrey Atkinson3, Horia I. Petrache1, Stephen R. Wassall1. 1 Department of Physics, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA, 2Department of Physics and Astronomy, Calvin College, Grand Rapids, MI, USA, 3Department of Chemistry, Brock University, St. Catharines, ON, Canada. Vitamin E (a-tocopherol) is a lipid-soluble antioxidant that has the role of protecting phospholipids from oxidation in membranes. A question that remains is how the low concentration of a-tocopherol found in whole cells can protect the relatively large concentration of polyunsaturated phospholipids found in membranes that are particularly vulnerable to oxidative attack. We hypothesize that a-tocopherol co-localizes with polyunsaturated phospholipids to optimize its role as an antioxidant. This project attempts to test this hypothesis by comparing the effect of a-tocopherol on the molecular organization of 1-palmitoyl-2-docosahexaenoyl-sn-glycerophosphatidylethanolamine (16:022:6PE, PDPE) and, as a monounsaturated control, 1-palmitoyl-2-oleoylsn-glycerophosphatidylethanolamine (16:0-18:1PE, POPE) in mixtures with sphingomyelin (SM). By solid-state 2H NMR spectroscopy, we directly observe order and phase behavior of POPE-d31 and PDPE-d31 (analogs of POPE and PDPE with a perdeuterated sn-1 chain) in the mixed membranes. In complementary X-ray diffraction and differential scanning calorimetry experiments we further probe phase behavior. The spectra observed for POPEd31 in POPE/SM/a-tocopherol (2:2:1 mol) reveal that a transition from gel to liquid crystalline phase is no longer apparent. At higher temperatures there is a superposition of two spectral components that we ascribe to a-tocopherol promoting a transition from lamellar to inverted hexagonal (HII) phase. Analysis of depaked spectra shows that order is increased by about 8 % and that the amount of HII phase increases with temperature, ranging from 7 (31 C) to 41 % (65 C). In mixed membranes where POPE-d31 is replaced by PDPE-d31, we shall investigate whether there is a greater tendency for a-tocopherol to increase order and destabilize bilayer structure for the polyunsaturated phospholipid. 2077-Pos Board B214 Molecular Dynamics Studies of Lipid I and Lipid II in Various of Lipid Bilayer Environments Seonghoon Kim, Wonpil Im. The University of Kansas, Lawrence, KS, USA. The peptidoglycan biosynthesis and assembly is needed for (both gramnegative and positive) bacterial cell wall formation. In the pathway, there are lipid-linked precursors such as lipid I (in the inner leaflet of the cytoplasmic membrane) and lipid II (in its outer leaflet). In addition, by peptidoglycan glycosyltransferase, one peptidoglycan unit from lipid II is transferred to the growing peptidoglycan oligomers attached to lipid II (lipid II-PG(x); x is the number of transferred peptidoglycan units). In this work, lipid I, lipid II-PG(0), lipid II-PG(2), lipid II-PG(5) of both gram-negative and positive bacteria are modeled and simulated in all-atom POPE/POPG (3:1; gram-negative membrane mimetics), POPE/POPG (1:3; gram-positive membrane mimetics), and POPC (neutral) bilayers. Presented and discussed are the structure and dynamics of the lipid-linked precursors and their dependence on bilayer types and vice versa. The preferred structures of these precursors are also discussed in terms of the reactions in (1) glycosyltransferase to add GlcNAc to lipid I, (2) peptidoglycan glycosyltransferase to add peptidoglycan units to lipid II-PG(x), and (3) transpeptidase that cross-links the peptidoglycan units. Tuesday, February 10, 2015 2078-Pos Board B215 Eicosapentaenoic Acid-Containing Membrane Domain Involved in Cell Division of a Cold-Adapted Bacterium Jun Kawamoto, Nobuyoshi Esaki, Tatsuo Kurihara. Institution for Chemical Research, Kyoto University, Uji, Kyoto, Japan. Long-chain omega-3 polyunsaturated fatty acids (PUFAs), such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), are found in organisms from bacteria to humans as the acyl group of phospholipids in the membrane. Many biophysical studies have been conducted on model membranes and revealed that PUFAs significantly alter the basic properties of lipid bilayers, such as the fluidity, acyl chain order, phase behavior, elastic compressibility, and permeability. However, despite accumulating information on the properties of the PUFA-containing bilayers, information on the physiological role of PUFAs and their molecular mode of action in living cells is very limited. In this study, we demonstrated a novel physiological function of EPA-containing phospholipids in a cold-adapted bacterium, Shewanella livingstonensis Ac10. We previously found that lack of EPA found at the sn-2 position of glycerophospholipids causes a defect in cell division of this strain. To study the localization of EPAcontaining phospholipids, we synthesized phospholipid probes labeled with a fluorescent group . A fluorescent probe in which EPA was bound to the glycerol backbone via an ester bond was found to be unsuitable for imaging because EPA was released from the probe by in vivo hydrolysis. To overcome this problem, we synthesized hydrolysis-resistant ether-type phospholipid probes. Using these probes, we found that the fluorescence localized between two nucleoids at the cell center during cell division when the cells were grown in the presence of the eicosapentaenyl group-containing probe, whereas this localization was not observed with the oleyl group-containing control probe. Thus, phospholipids containing an eicosapentaenyl group are specifically enriched at the cell division site. Formation of a membrane microdomain enriched in EPA-containing phospholipids at the nucleoid occlusion site probably facilitates cell division. 413a accommodating the variety of curvatures that would be required in the three dimensional arrangement of the lipid multilayers in skin, and for enabling mechanical or hydration induced strains without large curvature elastic costs. 2081-Pos Board B218 Hydration and Supramolecular Organization Studies of Lamellar Bodies in A549 Lung Cells using Laurdan Fluorescence Leonel S. Malacrida1,2, Soledad Astrada3, Mariela Bolatti3, Arturo Briva1, Luis A. Bagatolli4. 1 Fisiopatologı´a, Hospital de Clı´nicas, Montevideo, Uruguay, 2Unidad de Bioquı´mica y Proteo´mica Analiticas, Institut Pasteur Montevideo, Montevideo, Uruguay, 3Unidad de Biologı´a Celular, Institut Pasteur Montevideo, Montevideo, Uruguay, 4Membrane Biophysics and Biophotonics group/MEMPHYS -Department of Biochemistry and Molecular Biology, University of Southern Denmark, Odense, Denmark. The pulmonary surfactant is organized at the intracellular level in structures known as lamellar bodies (LBs). These subcellular acidic organelles have a variable size (aprox. 0.5-3 um) and based on the electron microscopy data it is proposed that they possess a concentrically membrane lamellar structure. Although the process of LB secretion by physiological inductors like ATP is well characterized, there is a lack of information related to their physical properties. Using the well known sensitivity of LAURDAN to membrane lateral organization we study LBs organization and dynamic properties of in vivo. We use an immortalized cell line from lung carcinoma, A549, which synthesize pulmonary surfactant and stores it as LBs. The fluorescence signal from LAURDAN was analyzed using the classical generalized polarization function and a newly method based on the Fourier transformation of the emission spectrum (called spectral phasor, Fereidouni-2012). The basic improvement of the spectral phasor is related to the Fourier transformation properties, which allow us the opportunity to decompose complex emission as a linear combination of single decay. Our results indicate that LBs have a highly packedmembrane structure with low extent of hydration in their core. This particular state is shifting to higher levels of fluidity with increased hydration levels at their periphery. The measured values of GP function indicates that the membrane in the core is in a gel like state (or liquid order state), though when the spectral data is analyzed a more complex system is identified. These results show higher lateral packing compared with DPPC vesicles but with a more heterogenic emission pattern. The interpretation opens the possibility to discuss the supramolecular organization and the role of water in these organelles. Fereidouni et al, Optics Express-2012 2079-Pos Board B216 Strong H-Bonds Form Bilayers: Ochromonas Danica is an Extreme Example Thomas H. Haines. Chemical Biology and Signal Receptors, Rockefeller University, New York, NY, USA. Anionic lipid bilayers provide the basic structure for nearly all living membranes. Anion-anion binding in head groups? I suggest a potent H-bond, a quantized Hbond (QHb). Biomembranes use glycerol-phospholipids with vicinal anionic phosphates. Maleate, 2-cis-succinate, is archetypical for a QHb. Its 2 carboxyls trap a proton between pH 2.1 to pH 6.2, contributing quantized bond energy to the molecule. Contrasting that with 2-trans-succinate (No H-bond) it contains an extra ~15kcal. The carboxyls are forced by the structure to share resonance with each other via the, ‘‘perhaps vibrating,’’ proton. Albeit in a narrow pH range, oleic acid and other unsaturated fatty acids do likewise. Here I report the structure of another anionic natural membrane, the chlorosulfolipid bilayer of Ochromonas danica. Its headgroups are QHb sulfates forced together by the hydrophobic chains of the bilayer. O. danica is an acidophilic (pH 4.3), fresh water alga in acid bogs. Its plasma membrane has two sets of single chain polar lipids: 2, 2, 11, 13, 15, 16, hexachloro-docosane-1,14-disulfate (>80%) and 2, 2, 12, 14, 16, 17-hexachloro-docosane-1, 15-disulfate, (<20%). Its C1-sulfates and C14/C15-sulfates are each arranged in sheets of sulfates within each monolayer. The latter sulfate sheet is made of sulfates separated by QHbs enclosed in a hydronium ion cage in the low dielectric. The chlorogroups bind hydronium ions so strongly that the CL-H3Oþ bond has 0.9 the strength of a covalent bond. They are arranged along the hydrocarbon chains to lead hydroniums toward the poly-sulfate cage. They enter the bilayer by chloro pair at C2, and are then passed down to the water cages. Both primary and secondary sulfate sheets are held together by QHbs, the strength of which prevent osmotic bursting. With phospholipid bilayers such Hbonds would be moving on a nanosecond timescale. 2082-Pos Board B219 Modulation of Phosphoinositide Monolayer Compressibilities by Physiological Levels of Ca2D Adolphe Kazadi Badiambile1,2, Martin B. Forstner2,3. 1 Physics ,Syracuse University, Syracuse, NY, USA, 2Syracuse Biomaterials Institute, Syracuse University, Syracuse, NY, USA, 3Physics, Physics,Syracuse University, Syracuse, NY, USA. Phosphoinositides (PIs) play a crucial role in many cellular processes that occur at the plasma membrane such as exocytosis and endocytosis. These processes not only locally enrich the membrane with PIs and are often accompanied by intracellular calcium release, but they also involve mechanical membrane deformations. Thus, the question arises how mechanical properties such as compressibility of highly negatively charged lipids such as phosphatidylinositol bisphosphate (PIP2) are modulated by physiological levels (0-1000nM) of Ca2þ ions. Using pure monolayers and a Langmuir film balance, we investigated the effect of bivalent ions on the compressibility of Phosphatidylinositol (PI), Phosphatidylinositol 4,5-bisphosphate (PIP2) , 1,2-dioleoyl-sn-glycero-3phospho-(10 -rac-glycerol) (DOPG)and 1-palmitoyl-2-oleoyl-sn-glycero-3phosphocholine (POPC). In addition, we also present a theoretical framework that describes the relationship between electrical surface potentials and compressibilities which shows good agreement with our experimental findings. 2080-Pos Board B217 Cholesterol Flip-Flop and Lack of Swelling in Stratum Corneum Lipid Bilayers Peter Olmsted1, Chinmay Das2, Massimo Noro3. 1 Physics, Georgetown University, Washington, DC, USA, 2Physics, University of Leeds, Leeds, United Kingdom, 3Unilever Research, Port Sunlight, United Kingdom. Atomistic simulations were performed on hydrated model lipid multilayers representative of the lipid matrix in the stratum corneum (the outermost layer of skin). We find that cholesterol transfers easily between adjacent leaflets belonging to the same bilayer via fast orientational diffusion (tumbling) in the interleaflet disordered region, while at the same time there is a large free energy cost against swelling. This fast flip-flop may play an important role in 2083-Pos Board B220 Proton Permeation through Extremophile-Inspired Lipid Membranes Thomas B.H. Schroeder1, Kathryn N. Haengel2, Mitchell A. Johnson2, Claire L. Wang3, Geoffray Leriche4, Takaoki Koyanagi4, Jerry Yang4, Michael Mayer5. 1 Chemical Engineering, University of Michigan, Ann Arbor, MI, USA, 2 Biomedical Engineering, University of Michigan, Ann Arbor, MI, USA, 3 Detroit Country Day School, Beverly Hills, MI, USA, 4Chemistry and Biochemistry, University of California, San Diego, San Diego, CA, USA, 5 Biomedical Engineering, Chemical Engineering, University of Michigan, Ann Arbor, MI, USA. The cell membranes of thermoacidophilic archaea contain lipids with unique structural motifs such as ether linkages, branched chains, and 414a Tuesday, February 10, 2015 membrane-spanning bipolar macrocycles that may allow the organisms to maintain the large pH gradient they require to survive. We investigated the relationship between the chemical structure of a number of lipids and the proton permeability of the membranes they form by using an optimized proton permeation assay performed on liposomes containing a fluorescent indicator dye. This work focuses on the effects of tethering on proton permeability and examines lipids with membrane-spanning chains of varied length and chemical structure (e.g. number and identity of rings). We discuss the results in the context of similar chemical groups and structures found in the cell membranes of extremophiles. 2084-Pos Board B221 Vibrational Spectroscopic Studies Probing Cardiolipin Containing Liposomes with and without Cytochrome C Bound to its Anionic Surface Dzmitry Malyshka, Leah Pandiscia, Reinhard Schweitzer-Stenner. Drexel University, Philadelphia, PA, USA. Cardiolipin is an important lipid on the inner mitochondrial membrane that interacts with cytochrome c, a protein in the electron transport chain that has been recently implicated to have a role in apoptosis. To properly characterize this interaction, the protonation state of cardiolipin needs to be identified. The literature has offered two opposing views with support for both the fully protonated and the semi-protonated state at physiological pH. Cardiolipin containing liposomes have long been used as model mitochondrial membranes. We measured the FTIR spectra of 1,10 2,20 -tetraoleyl cardiolipin (TOCL), 1,2-dioleyl-sn-glycero-3-phosphocholine (DOPC), and the more physiologically relevant 20% TOCL / 80% DOPC liposomes between the pH values of 2 and 11 in the region of 1000-1300 cm1. The spectra of DOPC liposomes were found to have no noticeable pH dependence. On the contrary, several bands of the spectra of TOCL containing liposomes increase or decrease in intensity at pH values below 4. These bands were assigned to normal modes with substantial contributions from PO4 and P¼O stretching modes, respectively, and they are diagnostic of the protonation state of the lipid. DFT based normal mode calculations revealed that the investigated spectral region is a superposition of bands assignable to collective CH deformation and PO4 stretching modes. This study suggests that the phosphate groups of the cardiolipin molecule are fully deprotonated at physiological pH. Recently, we performed resonance Raman studies to explore conformations and spin states of ferri- and ferrocytochrome c on 20% TOCL / 80% DOPC liposomes. We found that different binding sites give rise to different ligation states of the protein. 2085-Pos Board B222 Droplet Interface Bilayer as Cell Membrane Mimics: Water Permeability Studies Sunghee Lee. Iona College, New Rochelle, NY, USA. The process of water permeation across lipid membranes has significant implications for cellular physiology and homeostasis, and its study may lead to a greater understanding of the relationship between the structure of lipid bilayer and the role that lipid structure plays in water permeation. We have created a biomimetic artificial membrane, through contact of water droplets in an oil solution containing lipids. Using optical microscopy, we have measured water transport between droplets, as water moves from one droplet to another due to concentration difference. This was assessed as a function of lipid content, structure, and additives, such as cholesterol, which is an essential component of cell membrane. Our results show that cholesterol can increase the activation energy of water permeability several-fold, depending upon the structure of the lipid that makes up the bilayer, thus shedding light on how this singular sterol is vital to control of water movement. We demonstrate that the droplet interface bilayer can be employed as a convenient model membrane to rapidly explore subtle structural effects on bilayer water permeability. 2086-Pos Board B223 Comparison of Reactive Oxygen Species Production Activity and Binding Ability of Porphyrins in Cell Membrane Models Barnaba´s Bo¨cskei-Antal, Bianka Nagy, Szilvia Aniko´ To´th, Nikoletta Ko´sa, Istva´n Voszka, Gabriella Csı´k, Levente Here´nyi. Department of Biophysics and Radiation Biology, Semmelweis University, Budapest, Hungary. Photo dynamic therapy (PDT) is a widespread medical treatment based on the light-triggered generation of reactive oxygen species (ROS) by porphyrin derivatives. ROS may cause oxidative damage to membranes as well as to DNA and, in consequence, ultimately kill cells. Hence, the binding ability, the location within liposomes as simple cellular membrane models, and the ROS production ability of porphyrins are of outstanding interest. Earlier we determined the location of mesoporphyrin IX dimethyl ester (MPE) and its non-esterified form, mesoporphyrin IX dihydrochloride (MPCl) in small unilamellar vesicles (SUV) with fluorescence line narrowing spectroscopy (FLN). Here we investigated the production of ROS by the photosensitizers in the aqueous medium of the vesicles and in the lipid bilayer environment. The monocomponent vesicles were formed of various saturated phosphatidylcholines. The amount of generated oxygen radicals in the aqueous media was measured on the basis of the produced tri-iodide (I3-) from potassium iodide (KI) in the presence of molybdenum (MoO4) catalyst, which was followed by absorption spectrophotometry. The ROS in the lipophilic membranes and in near-membrane regions was measured with a dihydrorhodamine derivative by fluorescence spectroscopy. We observed in general that the binding ability of MPE is considerably higher than that of MPCl. In aqueous media (without liposomes) MPCl was highly effective in ROS formation whereas in case of MPE no similar effect was observed. Liposome-incorporated MPCl produced ROS in much higher amounts than the MPE in the aqueous medium of the liposomes. In near-membrane regions MPE produced ROS in the same amount as MPCl. Membrane Receptors and Signal Transduction III 2087-Pos Board B224 Investigating the Effect of Sodium and Voltage on d-Opioid Receptors Owen N. Vickery1, Daniel T. Baptista-Hon2, Daniel Seeliger3, Tim G. Hales2, Ulrich Zachariae1. 1 Divisions of Physics and Computational Biology, University of Dundee, Dundee, United Kingdom, 2Division of Neuroscience, University of Dundee, Dundee, United Kingdom, 3Boehringer Ingelheim GmbH&Co KG, Biberach an der Riss, Germany. G-protein-coupled receptors (GPCRs) are the largest superfamily of membrane proteins within the human genome. They participate in numerous physiological functions, including neuronal excitability and pain signalling. Owing to their functional and structural characteristics, they are excellent drug targets. In spite of their diversity, it is thought that GPCRs share a conserved pathway of signal transduction via conformational changes in their transmembrane (TM) domain. The full range of movements leading to activation, and their interaction with external factors, are however still incompletely understood. Many GPCRs are for instance modulated by sodium. The recent high-resolution crystal structure of the d-opioid receptor (dOR) provides detailed insight into the sodium binding site in the core of the TM domain [1]. In this work, we looked at the effect of sodium ions and transmembrane voltage on the flexibility and conformational changes of dORs. We applied a dual approach combining patch clamp electrophysiology and molecular dynamics simulations, in particular CompEl [2], to characterize the role of sodium in dOR. We studied the modulation of recombinant G-protein activated inwardly rectifying potassium (GIRK) channels by dORs in HEK cells, and simultaneously investigated the structure of dOR in double-bilayer, atomistic simulation systems under physiological and supra-physiological transmembrane electric fields. Our results implicate sodium as a key player in determining the global conformational ensemble of the dOR. [1] G. Fenalti et al., Nature 506, 191-196 (2014). [2] C. Kutzner et al., Biophys. J. 101, 809-817 (2011). 2088-Pos Board B225 Structure-Guided Discovery of Positive Allosteric Modulators of the Mu-Opioid Receptor Paola Bisignano1, Neil T. Burford2, Samuel W. Gerritz2, Andrew Alt2, Marta Filizola1. 1 Structural and Chemical Biology, Ichan School of Medicine at Mount Sinai, new york, NY, USA, 2Discovery, Bristol-Myers Squibb Company, Wallingford, CT, USA. The mu-opioid receptor (MOPr) continues to receive considerable attention in drug discovery efforts owing to its implication in pain management. Regretfully, activation of this receptor is also associated with significant adverse effects, including tolerance and abuse liability. In search for potent analgesics that are free from side effects, attention has recently shifted to allosteric modulators, that is, molecules that bind to (allosteric) sites on the receptor that are different from the orthosteric site recognized by endogenous agonists. The two recently reported positive allosteric modulators (PAMs) of the MOPr, i.e., BMS-986121 and BMS-986122, constitute the first example of such ligands. To facilitate their chemical optimization and/or discover additional PAMs of the MOPr, we searched for chemically similar compounds in the eMolecules database, and identified 1,336 molecules with a Tanimoto Tuesday, February 10, 2015 coefficient R 0.6 to either parent compound. The Schro¨dinger Suite 2014-2 was then used to prepare these molecules in a ready-to-dock format, including all tautomers and isomers. The available crystal structure of MOPr with a morphine replacing the crystallographic ligand at the orthosteric binding site was used for docking of all these compounds with Glide 6.3 using the XP scoring function. Following clustering of the top-ranked 10% compounds, forty-five representative ligands from the most populated clusters were selected for experimental testing based on visual inspection. As expected, the majority of small molecules tested showed PAM activity at the MOPr notwithstanding generally reduced activity values with respect to the parent compounds. One molecule stood out from the others given its comparable PAM activity to BMS-986121 and BMS-986122 in spite of a significantly different chemical scaffold. Additional studies are ongoing to explore the potential of this novel PAM of the MOPr as a lead for the development of new therapeutics. 2089-Pos Board B226 Binding Pockets and Poses of Allosteric Modulators of Opioid Receptors Identified by Metadynamics Yi Shang1, Holly R. Yeatman2, Neil Burford3, Kathryn Livingston4, Paola Bisignano1, John Traynor4, Andrew Alt3, Arthur Christopoulos2, Meritxell Canals2, Marta Filizola1. 1 Structural and Chemical Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA, 2Drug Discovery Biology, Monash Institute of Pharmaceutical Sciences and Monash University, Melbourne, Australia, 3 GPCR Lead Discovery & Optimization, Bristol-Myers Squibb Company, Wallingford, CT, USA, 4Department of Pharmacology, University of Michigan Medical School, Ann Arbor, MI, USA. As key targets for chronic pain, opioid receptors (ORs) are still at the forefront of drug discovery efforts. In the hunt for opioid analgesics that are free from adverse effects, recent high-throughput screening campaigns have focused on identifying allosteric modulators, that is molecules that bind non-conserved (allosteric) binding sites on the receptor, and modulate the potency and/or efficacy of ligands at the same (orthosteric) site as the endogenous agonist. Both positive and negative allosteric modulators (PAMs and NAMs) of m-OR and d-OR subtypes have recently been identified. While the recent crystal structures of ORs have revealed important details of ligand-receptor binding at the orthosteric site, both the location of allosteric sites on these receptors and the binding mode of opioid allosteric ligands are unknown. Here, we applied all-atom metadynamics to efficiently study the binding of a PAM and a NAM to the d-OR in the presence of the orthosteric agonist SNC-80 and an explicit lipid-water environment. The dynamics of the allosteric ligands was enhanced by biasing the potential acting on the ligand-receptor distance, and a contact map reflecting the ligand-receptor interaction. The resulting energy landscapes show two deep energy minima for both the simulated PAM and NAM, which correspond to the ligands acquiring specific binding poses within two different nearby receptor pockets defined by transmembrane helices TM1, TM2, and TM7. Notably, the ligand binding poses in the putative allosteric pocket that is closest to the orthosteric ligand overlap with the allosteric site predicted by the fragment-based mapping algorithm FTMAP. In spite of most ligand-receptor interactions being the same for the simulated PAM and NAM within this pocket, there are important differences that are being tested experimentally through mutagenesis to assess their functional role. 2090-Pos Board B227 Structural Dynamics and Energetics Underlying Allosteric Inactivation of a GPCR: Insights Gained from Site-Directed Fluorescence Labeling (SDFL) Studies of the Cannabinoid Receptor CB1 Jonathan Fay, David L. Farrens. Oregon Health and Science University, Portland, OR, USA. Biased signaling, signaling through non G-protein linked pathways, occurs in some GPCRs through mechanisms that are still not clear. Here we investigated how an allosteric ligand for the cannabinoid receptor, CB1, could induce such behavior, with the aim of identifying structural changes in the receptor and gaining mechanistic insights underlying these phenomena. The ligand, Org 27569, induces very unusual properties in CB1 - it increases agonist binding to the receptor, yet simultaneously decreases G-protein signaling while increasing biased signaling through beta-arrestin mediated pathways. Using classical pharmacological binding studies, we find that Org 27569 binds to a unique and separate site than traditional antagonists, and can act alone, without need for agonist co-binding. Similar analysis of constitutively activating and inactivating mutations in CB1, which shift the equilibrium between inactive (R) and active (R*) receptor conformations, indicate 415a that Org acts by modulating the amount of active R* species present. To determine the effect of Org on the structure of CB1, we carried out sitedirected fluorescence labeling (SDFL) studies. The SDFL results clearly show that Org induces structural changes in CB1 that are different than those caused by either antagonists or antagonists. While Org27569 blocks agonistinduced movements at TM6 (consistent with its ability to block G-protein signaling), it also enhances agonist-induced movements at H8, providing a clue to its ability to induce biased signaling. Together, these results help map the conformational dynamics involved in allosteric modulation and biased signaling in CB1. 2091-Pos Board B228 Optimization of Synthetically Novel Agonists of the Putative Cannabinoid Receptor, GPR55, using an Activated State Model Mary A. Lingerfelt1, Pingwei Zhao2, Lara Fakhouri1, Mary E. Abood2, Mitchell P. Croatt1, Patricia H. Reggio1. 1 Dept of Chem and Biochem, UNCG, Greensboro, NC, USA, 2Department of Anatomy and Cell Biology, Temple University, Philadelphia, PA, USA. The research presented details the creation of an activated state model of the putative cannabinoid receptor, GPR55, based on the crystal structure of CXCR4, a receptor with which GPR55 has high homology. Into this model, analogs of the high throughput screen compound hit, (E)-3-(2-methoxyphenyl)-N-((4-(N-methyl-N-phenylsulfamoyl)phenyl) carbamothioyl)acrylamide (CID1792197), were docked. Of particular interest was the synthetically novel GPR55 ligand, 6-(2-hydroxyethyl)-N-((4-(N-methyl-Nphenylsulfamoyl)phenyl)carbamothioyl)-2-napthamide, in that it is a highly bioactive analog of the parent molecule (CID1792197) . Both the novel ligand and its parent scaffold mimic the shape of lysophosphatidylinositol (LPI), the endogenous GPR55 ligand. Prior work in this lab established that the GPR55 binding pocket accommodates ligands with an inverted ‘‘L’’ or ‘‘T’’ shape, having long, thin profiles that can vertically penetrate deep in the receptor, while their broad upper regions occupy a horizontal binding pocket near the receptor’s extracellular loops. Key interactions currently known to exist between ligands and the GPR55 receptor are hydrogen bonding with K2.60(80), aromatic stacking with F3.33(102) and Van der Waals interactions with M3.36(105), F5.47(190)and L7.35(270). The results of the current project will facilitate the design and evaluation of third generation analogs as well as a radiolabeled, high affinity ligand that will allow for optimal characterization of the binding site interactions of the GPR55 receptor. [Support: NS077347-A1, DA023204 and DA021358] 2092-Pos Board B229 Signaling through Homomeric and Heteromeric Dopamine D2 and Cannabinoid Cb1 Receptors Guoqing Xiang. Physiology and Biophysics, Virginia Commonwealth University, RICHMOND, VA, USA. G protein-coupled receptors (GPCRs) play important roles in signal transduction and represent about half of the drug targets. Accumulating evidence suggests that GPCRs exist and function not only as homomers but as heteromers as well. Coimmunoprecipitation and fluorescence resonance energy transfer (FRET) results have provided evidence for heteromerization of D2-CB1 receptors in co-transfected cell lines. Besides, co-localization of CB1 and D2 receptors in the somata and dendrites of striato-pallidal GABA neurons and cortical-striatal glutamate terminals has been demonstrated. Studies have shown that homomeric D2 and CB1 receptors are Gi/o coupled able to inhibit upon stimulation the forskolin-induced adenylyl-cyclase activation leading to a decrease in cAMP levels. However, stimulation of the CB1 receptor upon the co-expression of D2-CB1 receptors in cell lines increased cAMP levels presumably because CB1 receptors switch their specificity to becoming Gs-coupled when complexed with the D2 receptor. In this study, we utilized Xenopus laevis oocytes as a heterologous expression system in which G protein-sensitive inwardly rectifying Kþ (GIRK) channels are co-expressed with GPCRs. Stimulation of GPCRs with specific ligands activates GIRK channels via the betagamma subunits of the G proteins (Gbg). The GIRK channel current is monitored by two-electrode voltage clamp (TEVC) to access receptor activity. We have found that stimulation of homomeric D2 and CB1 receptors increases the GIRK currents. This increase could be blocked by pertussis toxin (PTX) that specifically ADP-ribosylates Gi/o. Overexpression of Gs in Xenopus oocytes biases signaling to couple Gs coupled receptors to GIRK channels via Gbg. Using this approach, we demonstrated that neither the CB1 nor the D2 homomeric receptors can couple to Gs. We are in the process of testing whether the channel response of CB1 receptor ligands through the heteromeric D2-CB1 receptors proceeds via Gs. 416a Tuesday, February 10, 2015 2093-Pos Board B230 Pharmacological Implications of A2Ar-D2R Heteromerization and the Significance for Parkinson’s Disease Candice Hatcher-Solis, Diomedes E. Logothetis. Physiology and Biophysics, Virginia Commonwealth University, Richmond, VA, USA. Background: Recently, heteromeric GPCR complexes have become attractive targets for drug development since they exhibit distinct signaling and cell-specific localization from their homomeric counterparts. Yet, the effect of heteromerization on the pharmacology of many GPCR homomers remains unknown. We have undertaken the task to examine the effect of heteromerization on Gs signaling through the adenosine 2A receptor (A2AR) and Gi signaling through the dopamine receptor type 2 (D2R) because the A2AR-D2R heteromer is an emerging therapeutic target for Parkinson’s disease. Previous work suggests A2AR-D2R heteromerization induces reciprocal antagonism of homomeric signaling. Methods: We examined the effect of heteromerization on A2AR and D2R homomeric signaling using electrophysiology and the Xenopus oocyte heterologous expression system. GIRK channels were used as reporters for Gi signaling because activation leads to direct Gbeta-gamma-mediated stimulation of the GIRK current. We also coupled GIRK channels to Gs signaling by over expressing Gas. Our electrophysiological assay is innovative because it allows us to optimize the conditions of heteromerization. We anticipate that specific ligand combinations targeting the A2AR-D2R heteromer will be more efficacious than individual drug administration targeting A2AR or D2R homomers. Results: Preliminary data have demonstrated that heteromer formation decreases dopamine-elicited Gi signaling through the D2R and CGS-21680elicited Gs signaling through the A2AR. Furthermore, this reciprocal antagonism seemed to occur through a wide GPCR (cRNA) injection ratio. Currently, we are examining crosstalk by assessing whether A2AR agonists or inverse agonists will decrease or increase D2R-mediated Gi signaling through the A2AR-D2R heteromer. Modulation of Gs signaling through the A2AR by D2R ligands is also being examined. Conclusions: Our electrophysiological assay demonstrates A2AR-D2R heteromerization induces reciprocal antagonism reducing both A2AR and D2R signaling from that of homomeric levels. 2094-Pos Board B231 Structural Basis for the Allosteric Pharmacology of SB269652 in Dopamine D2 Receptor Mayako Michino1, Christopher J. Draper-Joyce2, Prashant Donthamsetti3,4, Jonathan A. Javitch3,4, J. Robert Lane2, Lei Shi1,5. 1 Department of Physiology and Biophysics, Weill Medical College of Cornell University, New York, NY, USA, 2Drug Discovery Biology, and Dept. of Pharmacology, Monash Institute of Pharmaceutical Sciences, Monash University (Parkville campus), Parkville, Australia, 3Departments of Psychiatry and Pharmacology, College of Physicians and Surgeons, Columbia University, New York, NY, USA, 4Division of Molecular Therapeutics, New York State Psychiatric Institute, New York, NY, USA, 5 Institute for Computational Biomedicine, Weill Medical College of Cornell University, New York, NY, USA. The dopamine D2-like receptors (D2R, D3R, and D4R), which belong to class A G protein-coupled receptors, are important targets for antipsychotics. In the development of novel antipsychotics to avoid adverse side effects, subtype-selective dopamine receptor ligands that bind divergent secondary binding pockets (SBP) in addition to the conserved orthosteric binding site (OBS) have been discovered. SB269652, an allosteric modulator of D2R, consists of a tetrahydroisoquinoline pharmacophore bound in the OBS and an indole-2-carboxamide moiety bound in a SBP between TM2 and TM7. This ligand was recently characterized to act allosterically by a novel mechanism involving a D2R homodimer whereby the bitopic binding mode of SB269652 in one protomer negatively modulates the binding of an orthosteric ligand, either an agonist or an inverse agonist, to the second protomer (Lane et al., Nature Chemical Biology, 2014). To characterize the structural basis for the allosteric effect of SB269652, we combined ligand modifications, complementary receptor mutagenesis, and computational analysis, to identify the key interactions between SB269652 and D2R and the correspondingly induced conformational changes that are critical to propagate the allosteric impact across the dimer interface. We specifically considered three pairs of SB269652 derivatives that are modified in the orthosteric or secondary pharmacophore, or the linker region - in each pair, the modifications either retain/improve or eliminate the allosteric property. Using molecular docking and dynamics simulations, we characterized the binding modes of these derivatives in D2R, either in a protomer alone or in a dimer context with an orthosteric ligand bound in the other protomer. The results of our comparative modeling and analysis highlight the importance of the receptor-ligand interaction formed in the SBP between Glu95(2.65) and the indole-2-carboxamide moiety, in modulating the conformational rearrangements of the dimer interface(s). 2095-Pos Board B232 Cross-Signaling between the Metabotropic Glutamate Receptor 2 and the Serotonin 2A Receptor in HEK-293 Cells Lia Baki1, Jason Younkin1, Jose Miguel Eltit1, Miguel Fribourg2, Amr Ellaithy1, Gyu Park1, Zhanna Vysotskaya1, Diomedes E. Logothetis1. 1 Physiology and Biophysics, Virginia Commonwealth University, Richmond, VA, USA, 2Neurology, Mount Sinai School of Medicine, New York, NY, USA. Atypical antipsychotic drugs targeting the 5-HT2A receptor (2AR) are widely used in the treatment of schizophrenia and psychosis. Recent studies point to a new class of potential antipsychotic drugs targeting the metabotropic glutamate receptor 2 (mGluR2). We recently reported that a heteromeric complex formed between these two receptors integrates the actions of serotonergic and glutamatergic drugs, modulating the balance between Gi and Gq signaling in a way that can predict the psychoactive properties of these drugs (Fribourg et al. 2011, Cell. 147(5):1011-23). The biological relevance of the mGluR2/2AR crosstalk was challenged by a more recent study in which co-expression of the two receptors in HEK-293 cells had no significant effect on either Gi or Gq signaling in response to several serotonergic and glutamatergic drugs (Delille et al., 2012, Neuropharmacology 62(7):2184-91). To address this controversy, we generated several clones of HEK-293 cells expressing different levels of the two receptors in the background of the G protein inwardly rectifying GIRK1/GIRK4 channel, which can serve as a reporter for both Gi and Gq signaling. Using the ratiometric calcium indicator Fura-2 for quantification of Gq signaling we identified clones showing various degrees of functional crosstalk between the two receptors (crosstalk positive clones) and clones that were crosstalk negative. Functional crosstalk correlated with the expression levels of the two receptors, as measured by real time RT-PCR and flowcytometry. GIRK channel activity, measured by electrophysiology, confirmed cross-signaling from both Gi and Gq sides in a crosstalk positive clone, but not a crosstalk negative one. In the later, a combination of ligands targeting both receptors was necessary in order to elicit functional crosstalk. We are currently testing positive allosteric modulators of the mGluR2 for their ability to suppress Gq signaling through 2AR. 2096-Pos Board B233 A Positive Allosteric Modulator of the Metabotropic Glutamate 2 Receptor Alters 5-HT2A Receptor Signaling in a Heteromeric Complex Amr Ellaithy, Jason Younkin, Lia Baki, Diomedes Logothetis. Virginia Commonwealth University, Richmond, VA, USA. Atypical antipsychotic drugs targeting the Gq-coupled serotonin 5-HT2A receptor (2AR) are widely used in the treatment of schizophrenia, whereas recent studies point to a new class of potential antipsychotic drugs targeting the Gicoupled metabotropic glutamate 2 receptor (mGluR2). A physical and functional interaction between the two receptors in cortical pyramidal neurons has been demonstrated. Using heterologously expressed receptors in Xenopus oocytes, we have previously shown that the heteromeric mGluR2/2AR complex integrates the actions of serotonergic and glutamatergic ligands, modulating the balance between Gi and Gq signaling in a way that can be used to predict the psychoactive properties of these drugs. Selective positive allosteric modulators (PAMs) of mGluR2 that bind to the transmembrane region of the receptor have shown efficacy in rodent models predictive of antipsychotic activity. Here we show that the mGluR2 PAM, LY487379 (or simply LY48) not only enhances glutamate-induced Gi signaling but also shows crosstalk by reducing 5-HT-induced Gq signaling. PAMs offer a promising therapeutic approach for the activation of GPCRs because they preserve the physiologic pattern of receptor signaling and are less likely to cause receptor desensitization and/or down regulation than synthetic agonists. 2097-Pos Board B234 Assembly and Cooperativity of Metabotropic Glutamate Receptors Josh Levitz, Reza Vafabakhsh, Shashank Bharill, Shashank Bharill, Ehud Y. Isacoff. UC Berkeley, Berkeley, CA, USA. Metabotropic glutamate receptors (mGluRs) are G-protein coupled receptors that are found throughout the nervous system where they respond to the major excitatory neurotransmitter, glutamate, to modulate synaptic transmission and plasticity via a variety of effectors. mGluRs are characterized by obligate dimerization and the presence of large extracellular ligand binding domains. In this study we used a combination of novel optical techniques to examine Tuesday, February 10, 2015 mGlu stoichiometry and to explore how these receptors cooperatively activate in response to ligand binding. We first used single molecule fluorescence photobleaching in live Xenopus oocytes and in receptors immobilized from mammalian cell lysate to map the dimer interfaces of homo and heterodimers. This work was complemented by experiments that use a covalently attached photoswitchable tethered ligand (PTL) that works as an agonist to analyze the cooperativity that arises from ligand binding to one or both subunits within a dimer. Finally, intersubunit FRET was used to analyze the conformational changes of mGluRs in order to gain an integrative view of receptor assembly, structural dynamics, and activation. 2098-Pos Board B235 Glycan-Based Connectivity Regulates the Hierarchical Organization of Membrane Receptors by Coupling their Micro- and Nano-Scale Lateral Mobility Juan A. Torreno-Pina1, Bruno Castro1, Alessandra Cambi2, Carlo Manzo1, Maria Garcia-Parajo1. 1 ICFO-Institute of Photonic Sciences, Castelldefels, Barcelona, Spain, 2 Research Institute of Molecular Life Sciences, Nijmegen, Netherlands. Glycan-protein interactions are emerging as important modulators of membrane protein organization and dynamics, regulating multiple cellular functions. In particular, it has been postulated that glycan-mediated interactions regulate surface residence time of glycoproteins and endocytosis. How this precisely occurs is poorly understood. We applied a combination of superresolution nanoscopy and single molecule-based approaches to study the role of glycan-based interactions on the dynamics of the glycosylated pathogen recognition receptor DC-SIGN, at the nano- and micrometer scale. We find that cell surface glycan-mediated interactions do not influence the nanoscale lateral organization of DC-SIGN in nanoclusters but restrict the mobility of the receptor to distinct micron-size membrane regions. These meso-scale regions are in turn enriched by the endocytic protein clathrin, thereby dynamically promoting DC-SIGN transient nano-scale arrest and interaction with clathrin. Disruption of glycan-based connectivity leads to larger membrane exploration, reduced clathrin interaction and compromised clathrindependent internalization of virus-like particles. Therefore, our work uncovers a novel mechanism through which glycan-protein interactions act as decisionmakers in fine-tuning membrane-related functions by dynamically coupling micro- and nanoscale receptor lateral mobility, thus adding a new layer of regulation to the hierarchical spatiotemporal organization of the cell membrane. 2099-Pos Board B236 The Role of Ligand Density in the Binding of Von Willebrand Factor by the Glycoprotein Ib-IX-V Complex in Platelets Zeinab Al-Rekabi1, Shirin Feghhi1, Nikita Taparia1, Adam D. Munday2,3, Wendy E. Thomas4, Jose A. Lopez2,3, Joachim P. Spatz5,6, Nathan J. Sniadecki1,4. 1 Department of Mechanical Engineering, University of Washington, Seattle, WA, USA, 2Puget Sound Blood Center, Seattle, WA, USA, 3Department of Medicine, Division of Hematology, University of Washington, Seattle, WA, USA, 4Department of Bioengineering, University of Washington, Seattle, WA, USA, 5Department of New Materials and Biosystems, Max Planck Institute for Intelligent Systems, Stuttgart, Germany, 6Department of Biophysical Chemistry, University of Heidelberg, Heidelberg, Germany. The initial arrest of platelets at a wound site requires the binding of glycoprotein Ib-IX-V complex (GPIb-IX-V) to the extracellular matrix protein von Willebrand factor (VWF). Increasing forces on these bonds increases the bond lifetime, known as a catch bond. Recently, we have shown that platelets are able to transmit cytoskeletal forces through the GPIba subunit of GPIb-IXV, which binds to the A1 domain of VWF. This provides an internal force on the bond to maintain adhesion in the absence of external forces. Integrin engagement and force transmission are known to require receptor clustering. We therefore investigate whether the clustering of GPIb-IX-V receptor is needed for the transmission of cytoskeletal forces to VWF. We first examined whether GPIb-IX-V clusters formed in spreading platelets. GPIba-positive punctate structures were visualized by confocal microscopy and correlated with dark regions imaged using interference reflection microscopy (IRM), indicating that GPIb-IX-V forms adhesive contacts that are similar in size to integrin-related focal adhesions in platelets. To investigate the effect of ligand density on platelet spreading, we prepared molecularly-defined adhesive ligand spots, which were separated 28 or 108 nm apart by nonadhesive regions using self-assembling gold nanoparticles. These nanoparticles were coated with A1 domain of VWF. When seeded on nanoparticles with 28 nm spacing, platelets appeared well-spread and formed punctate adhesions. However, when seeded on 108 nm spacing, platelet adhesion was significantly 417a reduced and those platelets that did adhere spread poorly. These findings demonstrate that an upper limit for ligand density exists where platelet adhesion and spreading are impeded. Investigating the effects of ligand spacing will facilitate an understanding of GPIba clustering in platelet adhesion and spreading, thus providing insight into thrombotic diseases and congenital bleeding disorders. 2100-Pos Board B237 Chelidonine Interferes with IL-6R/STAT3 Signaling in Uveal Melanoma Cells Istvan Csomos, Eniko Nizsaloczki, Gabriella Nagy, Laszlo Matyus, Andrea Bodnar. Department of Biophysics and Cell Biology, University of Debrecen, Debrecen, Hungary. There is increasing evidence suggesting the importance of IL-6 in oncogenesis: it stimulates tumor cells proliferation and promotes cell survival through the inhibition of apoptosis. IL-6 acts on a receptor complex consisting of the cytokine-specific IL-6Ra chain and the signal-transducing gp130 subunit. Binding of IL-6 to IL-6Ra induces dimerization of gp130 which initiates multiple signaling cascades, including STAT3 activation. Chelidonine, the major alkaloid component of C. majus, provokes cell death in a variety of tumor cells, possibly through the antiapoptotic Bcl-2 protein. Expression of Bcl-2 is upregulated by STAT3 activation, which is thought to be responsible for IL-6-mediated survival of tumor cells. Herein we aimed to study the effect of chelidonine on the viability of human uveal melanoma cells as wells as its interference with the IL-6R/STAT3 signaling pathway. Antiproliferative and cell death-inducing effects of chelidonine were assessed by flow cytometry. The apoptotic potential of chelidonine was followed by DNA fragmentation and PI exclusion/annexin V binding assays. Expression of STAT3, Bcl-2 and IL-6Ra and the efficiency of STAT3 activation was also studied by flow cytometry. Combined analysis of cell death experiments revealed chelidonine-induced apoptosis of UM cells. Moreover, alkaloid treatment also resulted in necrotic cell death. Pretreatment of cells even with sublethal doses of chelidonine led to the appearance of a subpopulation with abrogated STAT3 activation upon IL-6 stimulation and modified Bcl-2 expression levels. We detected cells with reduced expression of STAT3 and IL-6Ra; however, the amount of these cells was significantly lower than that of cells with abolished STAT3 signaling. According to our results chelidonine exerts its effect via a STAT3-dependent mechanism. Our findings imply the possible use of chelidonine in cancer therapy: it can either provoke cell death or weaken the antiapoptotic machinery of tumor cells fueled by IL-6. 2101-Pos Board B238 The Site of Arachidonic Acid Release Drives Calcium Dynamics in b-Cells Dmytro A. Yushchenko, Andre´ Nadler, Rainer Mueller, Frank Stein, Gurleen Khandpur, Suihan Feng, Carsten Schultz. Cell Biology and Biophysics, The European Molecular Biology Laboratory (EMBL), Heidelberg, Germany. Insulin is the blood glucose-lowering hormone essential for glucose homeostasis. b-Cells secrete insulin in pulses in response to glucose. Loss of the oscillatory nature of insulin secretion is associated with the development of type II diabetes. Calcium is the most important trigger of insulin release even though the exact mechanism of insulin secretion regulation by calcium is elusive. Arachidonic acid (AA) is an essential signalling molecule involved in regulation of physiological functions of many cell types. In insulin secreting pancreatic b-cells AA was shown to modulate calcium levels and as result to trigger insulin secretion. However, the exact interplay between AA signalling and insulin secretion is still a matter of speculation. In the present work we investigated the difference of AA action at the internal membranes (IMs) and the plasma membrane (PM) of b-cells. To perform this study, we developed a caging group which permits localization, visualization and quantitative photorelease of AA exclusively on the PM of living cells. We applied it in combination with a previously reported caging group used to release AA on the IMs. We found that the release of AA on the PM and the IMs leads to a significantly different modulation of intracellular calcium dynamics. Uncaging of AA on the PM induces calcium oscillations in non-oscillating cells and increases the duration of calcium transients in oscillating cells, leading to overall higher calcium levels. Release of AA on the IM results in transiently or permanently diminished calcium oscillations in b-cells and lower average calcium levels. We attribute the observed effects to direct action of AA on Arachidonic acid Regulated Calcium (ARC) channels localized on the PM and potentially a negative feedback mechanism triggered by higher levels of AA at the internal membranes. 418a Tuesday, February 10, 2015 2102-Pos Board B239 Multi-Scale Linkages between Single-Molecule Integrin Dynamics and Cell Protrusion Khuloud Jaqaman1, James A. Galbraith2, Michael Davidson3, Gaudenz Danuser1, Catherine G. Galbraith2. 1 UT Southwestern Medical Center, Dallas, TX, USA, 2Oregon Health and Science University, Portland, OR, USA, 3Florida State University, Tallahasee, FL, USA. Recent advances in light microscopy permit the development of cytoarchitectural blueprints with single-molecule resolution. It is now conceptually possible to relate the organization of the molecular machinery to cellular function. However, the disparity between the spatial and temporal scales of molecular and cellular behaviors poses substantial challenges in deriving these relationships. New approaches are required to integrate discrete single-molecule behavior with continuous cellular-level processes. Here, we combined intercalated molecular and cellular imaging with an analytical framework to reveal the roles that individual integrin molecules play in the process of initiating cell adhesion and migration. Despite the stochasticity of molecular and cellular behaviors, our data uncovered ‘rules’ of molecular organization for the components of a functional nascent adhesion, namely avb3-integrin, talin and actin, as well as the relationship between the local molecular organization of integrins and cell edge movement. We found that integrins exhibit a basal spatial gradient, with highest density and slowest mobility at the cell edge. In preparation for protrusion, this gradient transiently and locally increases, indicative of the initiation of nascent adhesions prior to cell edge protrusion. Moreover, the molecular behavior of talin, actin and b3 integrin mutants revealed that talin binding regulates integrin mobility, while integrin density modulation is primarily dependent on actin. These data establish a multi-scale linkage between characteristic variations in discrete stochastic molecular behaviors and continuum cellular behaviors. 2103-Pos Board B240 Weak Ergodicity Breaking of Membrane Receptor Motion Stemming from Random Diffusivity Carlo Manzo1, Juan A. Torreno-Pina1, Pietro Massignan1, Gerald J. Lapeyre, Jr.1, Maciej Lewenstein1,2, Maria F. Garcia-Parajo1,2. 1 ICFO, Castelldefels, Spain, 2ICREA, Barcelona, Spain. The application of techniques such as fluorescence correlation spectroscopy and single particle tracking to the plasma membrane of living cells continuously evidences subdiffusive motion of proteins and lipids. This anomalous motion is generally associated to the interplay of molecular crowding, diffusion barriers and specific interactions. Addressing the cause of subdiffusion is essential for understanding molecular mechanisms underlying cellular function, such as target search, kinetics of transport-limited reactions, trafficking and signalling. Recently, the subdiffusive motion of some cellular and membrane components has been associated to weak ergodicity breaking and attributed to transient immobilization caused by interactions with microtubules or actin cytoskeleton. These works have opened new questions about the role of nonergodic subdiffusion in the dynamics of living systems. By means of single particle tracking experiments, we show that the dynamics of DC-SIGN, a transmembrane receptor with unique pathogen recognition capabilities, also reveals subdiffusion with signatures of weak ergodicity breaking and aging. In contrast to other biological systems, we demonstrate that DC-SIGN nonergodic subdiffusion cannot be explained by transient immobilization, but rather is compatible with dynamical heterogeneity induced by spatiotemporal changes of diffusivity. While nonergodic diffusion due to long-lived immobilization events can be understood within the framework of continuous-time random walk, our experimental data are accurately interpreted through a new theoretical model describing anomalous transport in biological systems and complex media. A comprehensive analysis of mutated forms of the receptor allowed us to establish a link between receptor molecular structure, nonergodic diffusion and function. Our results underscore the role of spatiotemporal disorder in the dynamics of cell membrane receptors. In addition, they have broad implications to other heterogeneous systems where the occurrence of nonergodicity remains unexplored. 2104-Pos Board B241 Restricted Mobility of TonB and FepA in E. coli Membranes Yoriko Lill1, Lorne D. Jordan2, Chuck R. Smallwood3, Salete M. Newton2, Phillip E. Klebba2, Ken Ritchie1. 1 Purdue University, West Lafayette, IN, USA, 2Kansas State University, Manhattan, KS, USA, 3University of Oklahoma, Norman, OK, USA. Nutrient uptake in Escherichia coli requires transport across the cell’s lipopolysaccharide-rich outer membrane, passage through the peptidoglycan layer containing periplasmic space and finally transport across the cell’s inner cytoplasmic membrane. In many cases, the first step in this process involves transit through a class of beta-barrel proteins in the outer membrane known as TonB-dependent transporters (TBDTs). This uptake requires interaction between the TBDTs and the cytoplasmic membrane protein, TonB. TonB, in complex with ExbB and ExbD, is thought to derive energy from protonmotive force of the cytoplasmic membrane to be transduced to the outer membrane TBDTs for transport. Here, we have investigated the mobility of TonB in the inner membrane and FepA, a TBDT responsible for transport of the iron chelating siderophore enterobactin (FeEnt), in the outer membrane of E. coli at the single molecule level. We find that FepA is located throughout the outer membrane while TonB is found excluded from the inner membrane’s polar regions. Both proteins are observed to undergo free diffusion in the absence of FeEnt within confining domains in their respective membranes (a domain radius of 130 nm for FepA and 240 nm for TonB). Effects of depolarization of the inner membrane, deletion of ExbB/D and presence of FeEnt will be discussed in term of interactions between the proteins. 2105-Pos Board B242 Cryo-Electron Tomography and Computer Simulations Reveal Distinct CheA Kinase Conformation in Bacterial Chemotaxis Signaling Receptor Complex Benjamin A. Himes1, C. Keith Cassidy2, Jun Ma1, Frances Joan D. Alvarez1, Juan R. Perilla2, Gongpu Zhao1, Klaus Schulten2, Peijun Zhang1. 1 Structural Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA, 2Department of Physics and Beckman Institute, University of Illinois at Urbana-Champaign, Urbana, IL, USA. Improved sample preparation, data collection, and image processing pipelines have ushered in a new era in cryo-Electron Tomography (CryoET). Electron density maps of large, conformationally heterogeneous assemblies can be determined at subnanometer resolutions and coupled with high resolution structural information and large scale molecular dynamics simulation to gain atomistic understanding of large and complex cellular machinery that is otherwise unattainable. We use such an integrative approach to investigate the signal transduction in bacterial chemotaxis arrays - large transmembrane protein complex assemblies responsible for modulating the motility of bacteria. These arrays are responsible, in part for appropriately biasing the cell’s inherent random walk, and also retaining the memory of recent states of the system. Despite recent advances, fundamental questions on how the components of the system work together to transduce the signal from membrane receptor to kinase molecule, and how the individual signaling unit works cohesively to achieve a large gain in signaling remain unanswered. We devised a novel in vitro reconstitution system to build extended signaling arrays on a lipid monolayer using purified protein components, providing an unprecedented number of signaling units in a thin layer (~150-200nm) ideal for cryoET. The individual subvolumes from 3D tomograms are extracted, aligned, classified and averaged, resulting in a 3D density map of the basic chemotaxis signaling unit comprising the chemoreceptor, the histidine kinase CheA and the cofactor CheW. The density map reveals an unexpected asymmetry among three kinase CheA dimers, indicating distinct CheA conformations within the unit. Large scale all-atom molecular dynamics simulations further point to the specific side chain contacts that mediate the transition of CheA conformations, which we show, through biochemical assays, to fully abrogate chemotactic function if disrupted. 2106-Pos Board B243 Tracking Ceacam1 Interactions and Dynamics with homo-FRET and Image Correlation Techniques Amy M. Won1, Scott D. Gray-Owen2, Christopher M. Yip1. 1 Institute of Biomaterials and Biomedical Engineering, University of Toronto, Toronto, ON, Canada, 2Molecular Genetics, University of Toronto, Toronto, ON, Canada. The carcinoembryonic antigen (CEA) family plays an important role in numerous normal and pathogenic processes related to cellular growth and differentiation. Crucial roles of various CEA-family members are well established while the effect of carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1) on immune-cell function has only recently been recognized. CEACAM1 is known to exist in both monomeric and dimeric states that are heterogeneously distributed at the cell surface. However, which form participates in cell-cell interactions and its related dynamics remain unknown. We report here details of CEACAM1’s spatial distribution, oligomeric state, diffusional characteristics and dynamics as determined by live cell homo-fluorescence energy resonance transfer (homo-FRET) microscopy and imaging total internal reflection fluorescence correlation spectroscopy (ITIR-FCS). Tuesday, February 10, 2015 2107-Pos Board B244 Molecular Mechanism Associated to the Offspring’s Cognitive Impairment due to Maternal Thyroid Hormones Deficiency during Gestation Maria Cecilia Opazo1,2, Luis Venegas1,2, Pablo Cisternas1,2, Eduardo Albornoz1, Enzo Seguel1,2, Susan Bueno2,3, Alexis Kalergis2,3, Claudia Riedel1,2. 1 Universidad Andres Bello, Santiago, Chile, 2Milleniun Institute on Immunology and Immunotherapy, Santiago, Chile, 3Pontificia Universidad Cato´lica de Chile, Santiago, Chile. Maternal thyroid hormones (MTH) have a key role during fetal development. MTH deficiency during gestation can cause cognitive alterations in the offspring. Two main MTH deficiencies are described: hypothyroidism (low T4, high TSH) and hypothyroxinemia (low T4, normal TSH and T3). The molecular role of MTH has not been yet fully elucidated. THs exert a nuclear genomic effect by inducing the expression of target genes. T3 can bind to thyroid hormone receptors (TRs), promoting their translocation to the nucleus and modifying the expression of TH-responsive genes. Here, we analize in vivo and in vitro the expression of TR, TH transporters and key proteins associated to the molecular mechanism of TH, in brain cortex and hippocampus of adult offspring gestated under MTH deficiency. At functional level, long tem potentiation (LTP) was evaluated in vivo and chemical LTP (chLTP) was evaluated in vitro. An alteration into TR and transporters expression levels was observed in cortex and hippocampus together with an alteration of both LTP and chLTP. This work contributes to elucidate the molecular mechanism associated to the MTH role in the progeny CNS development. 2108-Pos Board B245 P2X4 Forms ATP-Activated Channels on Lysosomal Membranes Regulated by Luminal pH and SLC17A9 Proteins Xianping Dong. Physiology and Biophysics, Dalhousie University, Halifax, NS, Canada. P2X receptors are commonly known as plasma membrane cation channels involved in a wide variety of cell functions. Recently, it was shown that in addition to plasma membrane expression, mammalian P2X4 was also localized intracellularly in lysosomes. However, it was not clear whether the lysosomal P2X4 receptors function as channels and how they are regulated. In this study, we show that both P2X4 and its natural ligand, ATP, are enriched in lysosomes of mammalian cells. By directly recording membrane currents from enlarged lysosomal vacuoles, we demonstrate that lysosomal P2X4 proteins form channels activated by ATP from the luminal side in a pH dependent manner. While the acidic pH at the luminal side inhibits P2X4 activity, increasing the luminal pH in the presence of ATP causes P2X4 activation. We further show that SLC17A9 (solute carrier family 17 member 9), also known as VNUT (vesicular nucleotide transporter), functions as an ATP transporter in lysosomes where it transports ATP into lysosomes to activate P2X4 channels. Taken together, our data suggest that P2X4 acts as a lysosomal channel regulated by a number of factors including luminal ATP, luminal pH, and membrane SLC17A9. Such regulation may be important for lysosome physiology. Excitation-Contraction Coupling II 2109-Pos Board B246 Identification of Major FKBP12 Binding Determinants in RyR1 Razvan L. Cornea1, Filip Van Petegem2, James D. Fessenden3. 1 Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, USA, 2Biochemistry and Molecular Biology, University of Vancouver, Vancouver, BC, Canada, 3Anesthesia, Brigham and Women’s Hospital, Boston, MA, USA. We used site-directed mutagenesis and labeling of the type 1 ryanodine receptor (RyR1) recombinantly expressed in HEK-293T cells to identify RyR1 residues that interact with FKBP12, a constitutively-bound protein that promotes the closed state of the RyR1 calcium channel. While the location of bound FKBP on the RyR1 cryo electron microscopic (EM) map is known, the RyR1 sequence elements forming the FKBP binding site are poorly understood. Previously, we have shown that a His10 tag inserted at RyR1 position 620, adjacent to the N-terminal ‘‘ABC’’ domains, abolished FKBP binding. Here, we used structural modeling to predict loop regions in downstream domains, and then used these loops for His10 insertion sites. Fluorescent FKBP (F-FKBP) binding was then assessed in functional, full-length RyR1 containing these His10 tags. We determined that 419a insertion of His10 tags in specific loops could completely abolish FFKBP binding, even though RyR1 calcium release activity was unaffected. Scrambling the amino acid sequence of the loops identified using His10 tags also eliminated F-FKBP binding. The effects of disrupting these loops on F-FKBP binding were far more pronounced compared to previously established FKBP binding motifs, such as the VP dipeptide at RyR1 position 2461 and the PKA phosphorylation site at position 2843 within the PKA/ CaMKII phosphorylation loop of RyR1. Thus, our results identify novel major FKBP binding determinants in RyR1. Supported by NIH and Canadian grants R01AR059126 (to JDF) R01HL092097 (to RLC), and CIHR 259009 (to FVP). 2110-Pos Board B247 Genetic Deletion of Fkbp12.6 Accelerates Cardiac Aging in Mice Guangju Ji. Institute of Biophysics, CAS, Beijing, China. Aims: Cardiac aging is one of the main causes of heart dysfunction and other cardiac diseases. FK506 binding protein 12.6 (FKBP12.6) modulates Ca2þ release from the sarcoplasmic reticulum in cardiomyocytes. Previous studies implied that genetic deletion of FKBP12.6 leads to cardiac aging related phenotypes. However, the role and mechanism of FKBP12.6 in the process of cardiac aging remain unclear. Methods and Results: To assess whether FKBP12.6 is involved in cardiac aging and age-related heart dysfunction, FKBP12.6 knockout (KO) and control littermate mice were used throughout the study. We found a significant association between deletion of FKBP12.6 and cardiac aging. Indeed, aged FKBP12.6 KO mice exhibited markedly impaired cardiac function compared to wild-type. Strikingly, FKBP12.6 deletion resulted also in increased levels of cell cycle inhibitors p16 and p19, augmented cardiac fibrosis, cell death, and shorter telomeres. Finally, we demonstrated that FKBP12.6 deletion enhanced insulin signaling in the hearts, resulting in AKT phosphorylation, augmented mTOR activity, and impaired autophagy. Conclusions: Taken together, these results identify that genetic deletion of FKBP12.6 promotes cardiac aging and indicate that the activation of the AKT/mTOR pathway plays a mechanistic role in the process. 2111-Pos Board B248 Properties of Atrial Myocyte Calcium Handling in Canine Model of Chronic Heart Failure Andriy Belevych1, Hsiang-Ting Ho1, Qing Lou1, Lucia Brunello1, Ingrid Bonilla2, Karsten Schober3, Kent Mowrey4, Raul Weiss5, Cynthia A. Carnes2, Sandor Gyorke1. 1 Physiology & Cell Biology, OSU, Columbus, OH, USA, 2College of Pharmacy, OSU, Columbus, OH, USA, 3College of Veterinary Medicine, OSU, Columbus, OH, USA, 4St. Jude Medical, St .Paul, MN, USA, 5Division of Cardiology, OSUMC, Columbus, OH, USA. Weakened atrial contractility is a common comorbidity in heart failure (HF) that increases severity of HF symptoms. The role of intracellular calcium (Ca) handling remodeling in pathogenesis of ventricular contractile failure has been well established. However, the relationship between Ca signaling and atrial dysfunction in HF is not well recognized. We studied intracellular Ca handling in atrial myocytes isolated from control dogs and dogs with tachypacing-induced chronic HF. Ca transients and cellular shortening were reduced, while sarcoplasmic reticulum (SR) Ca content was increased in field-stimulated myocytes from HF compared to controls. Both control and HF myocytes have scarce t-tubule density, and Ca transients originated predominantly at the periphery of myocytes in both groups. Diminished Ca transients in HF myocytes were associated with decreased peripheral Ca release and impaired propagation of Ca signal from sarcolemma to the central areas of the myocyte. Peak density of the L-type Ca currents and gain of excitation-contraction coupling were reduced in voltage-clamped HF myocytes compared to control myocytes. Analysis of decay of caffeine- and electrically-induced Ca transients suggests reduced Na/Ca exchanger activity and increased SR Ca ATPase (SERCA) activity in HF myocytes. SERCA inhibition with cyclopiazonic acid did not improve centripetal propagation of Ca signal in HF myocytes. In contrast, L-type Ca channel agonist BayK 8644 increased peripheral Ca release, improved centripetal Ca wave propagation and restored shortening of HF myocytes to control levels. We conclude that defective activation and impaired propagation of the SR Ca release in atrial myocytes contribute to compromised atrial contractility in chronic HF. Our results suggest L-type Ca channels as a therapeutic target for treatment of atrial contractile dysfunction in HF. 420a Tuesday, February 10, 2015 2112-Pos Board B249 Effect of Hypertrophic Calcium Signals and Altered ExcitationContraction Coupling on the Calcineurin-NFAT Pathway Joseph L. Greenstein, Tejas Mehta, Raimond L. Winslow. Institute for Computational Medicine, Johns Hopkins University, Baltimore, MD, USA. Many questions remain unanswered as to how calcium (Ca2þ) signals regulate the Calcineurin (CaN)- Nuclear Factor of Activated T-cells (NFAT) signaling pathway. The CaN-NFAT cascade is one of the key pathways regulating the development of cardiac hypertrophy, a condition which is recognized as a leading risk factor for heart failure. In addition, the effect of certain functional modifications in proteins involved in excitation-contraction coupling (ECC) on cardiac hypertrophy is still not well understood. In this work, we develop a mathematical model for the CaN-NFAT pathway and incorporate it into an integrative computational model of the ventricular myocyte to better understand the relationship between Ca2þ signals, CaN-NFAT, and hypertrophy. Model results suggest the frequency of calcium transients (i.e. heart rate) may be a primary determinant of hypertrophic signaling, whereas diastolic Ca2þ level and inositol trisphosphate (IP3)-mediated changes in nuclear Ca2þ level appear to play less of a role. Furthermore, alterations in the function of ECC proteins as may occur under conditions of stress, heart disease, and/or therapeutic intervention were investigated. Results indicate L-type Ca2þ channel overexpression increases NFAT activity, while overexpression of the sarcoplasmic reticulum Ca2þ ATPase, which mimics the functional effect of therapeutic SERCA gene transfer, has a relatively minor effect on NFAT activity. Therefore, an important prediction of this model is that enhancement of SERCA function is not predicted to promote the hypertrophic response, and it therefore may be possible to treat both pathological hypertrophy and SR dysfunction simultaneously. 2113-Pos Board B250 EC Coupling for Muscle Aficionados: Abnormal Contraction and Disrupted Excitability in Some Enzymatically Dissociated Skeletal Muscle Fibers Camilo Vanegas, Martin F. Schneider, Erick O. Herna´ndez-Ochoa. Department of Biochemistry and Molecular Biology, University of Maryland, Baltimore, MD, USA. Enzymatically dissociated skeletal muscle fibers are routinely used in studies of the excitation-contraction coupling (ECC) process. Here we show that skeletal muscle fibers enzymatically dissociated and cultured for 1-2 days are in general useful for experimentation; however a subset of fibers, despite their normal appearance, exhibit abnormal contractility and calcium transient alterations. We monitored the contractile response of fibers to electric field stimulation using transmitted light microscopy, and the spatial distribution of the calcium transients using Rhod-2, a non-ratiometric calcium indicator, and ultra-fast confocal microscopy. A variable fraction of fibers expressed abnormal ECC properties; they were unable to contract homogenously in response to electrical stimuli, and most notably exhibited alternating local contractions at fiber ends in response to stimuli of alternating polarity. These alternating (ALT) response fibers exhibit minor gross morphological differences from normal (WT) fibers, which contract uniformly and respond to both polarity electrical pulses. In some ALT fibers, di-8-anepps staining shows a partial disruption of the T-tubule network at the center of the fibers. This might indicate that T-tubule network disruption accompanies changes in excitability observed in ALT fibers. We hypothesize that changes in the function and/or expression of voltage gated channels, responsible for action potential generation, or voltage gated calcium channels, essential for ECC, could explain our results. Regardless of the mechanism(s) responsible for lack of excitability, we encourage the careful monitoring of the contractile response of the fiber (and if available, the evaluation of the calcium transients) to determine that normal behavior of skeletal muscle fibers be implemented when choosing fibers for physiological experiments, and recommend avoiding the use of muscle fibers that display only local contractile activity to alternating polarity. Supported by NIH-NIAMS award R37-AR055099. 2114-Pos Board B251 Excitation-Contraction Coupling in Human Extraocular Muscles:There is more than Meets the Eye Marijana Sekulic-Jablanovic1, Anja Palmowski-Wolfe2, Francesco Zorzato1,3, Susan Treves1,4. 1 Departments of Research and Anaesthesia, Basel University Hospital, Basel, Switzerland, 2Eye hospital, Basel University Hospital, Basel, Switzerland, 3 Life sciences, University of Ferrara, Ferrara, Italy, 4Life Sciences, University of Ferrara, Ferrara, Italy. Excitation-contraction coupling is the physiological mechanism whereby an electrical signal detected by the dihydropyridine receptor, is converted into an increase in [Ca2þ], via activation of ryanodine receptors. Mutations in RYR1, the gene encoding ryanodine receptor 1, are the underlying cause of various congenital myopathies including Central Core Disease, Multiminicore disease, some forms of Centronucleal myopathy and Congenital Fiber Type disproportion. Interestingly, patients with recessive but not dominant RYR1 mutations show a significant reduction of ryanodine receptor protein in muscle biopsies as well as ophthalmoplegia. In order to understand why the patients with recessive RYR1 mutations specifically showed involvement of the extraocular muscles, we investigated the excitation-contraction coupling machinery in biopsies obtained from ‘‘normal’’ patients undergoing resection eye surgery. Our results show that the major proteins involved in skeletal muscle excitationcontraction coupling are expressed differently in human extraocular muscles compared to leg muscles. In particular the transcripts encoding ryanodine receptor 3, cardiac calsequestrin and the alfa 1 subunit of the cardiac dihydropyridine receptor were upregulated by at least 100 fold, whereas ryanodine receptor 1 and the alfa 1 subunit of the skeletal dihydropyridine receptor were reduced 10 fold. Myotubes obtained from extraocular muscle biopsies also exhibited changes in their calcium homeostasis, and particularly the resting [Ca2þ] was lower and the depolarization induced Ca2þ influx was 3 fold higher compared to that observed in leg muscle-derived myotubes. These results indicate that extraocular muscles have a different mode of calcium handling; moreover, the presence of ophthalmoplegia in patients with recessive RYR1 mutations is most likely due to the lower endogenous levels of RyR1 expressed by the extraocular muscles. 2115-Pos Board B252 The Calcium-Activated Chloride Channel in Zebrafish Skeletal Muscle is Activated during Excitation-Contraction Coupling Shu Fun Josephine Ng, Anamika Dayal, Manfred Grabner. Medical University Innsbruck, Innsbruck, Austria. Ca2þ-activated Cl- channels (CaCC) are expressed in various tissues and play important roles in numerous physiological functions such as epithelial secretion, olfactory and sensory transduction, cardiac excitability, and smooth muscle contraction. Even though CaCC mRNA was identified in human skeletal muscle (Huang et al., 2006), no CaCC conductance has been reported till date. Surprisingly, we found robust Ca2þ-activated Cl- currents with current amplitudes of >100pA/pF at þ80mV membrane potential in zebrafish skeletal muscle cells. Immunocytochemistry and subtype-specific CaCC current blockers allowed us to identify Ano1 (TMEM16A) as the channel protein responsible for this massive Cl- influx in zebrafish skeletal myotube. Wholecell patch-clamp recordings revealed that this CaCC current is outwardly rectifying at sub-maximal Ca2þ levels and shows a linear current-voltage relationship at high [Ca2þ]. Interestingly, this robust CaCC current can only be observed in wild-type zebrafish myotubes, which display intact SR Ca2þ release during excitation-contraction (EC) coupling. In contrast, the CaV1.1 b1-null zebrafish mutant relaxed, lacking the SR Ca2þ release, displayed no CaCC current. Thus, the CaCC current through Ano1 is activated by SR Ca2þ release during EC coupling. CaCC activation during EC coupling is close to maximum regarding Cl- influx, as seen in Ca2þ-dependence experiments. Furthermore, we observed different CaCC current properties in the superficial slow and deep fast skeletal musculature. Further studies, to test if the CaCC current in zebrafish skeletal muscle plays a role in shaping the action potential by shortening the repolarization phase and thus allowing faster muscle contraction, are on the way. Supported by FWF P23229 and W1101-B12 Muscle Regulation 2116-Pos Board B253 Proteins in Striated Muscles that Transcribed from the Contiguous Region of Connectin Gene Akira Hanashima1, Naruki Sato2, Sumiko Kimura2, Takashi Sakurai1, Takashi Murayama1. 1 Pharmacology, Juntendo University, Tokyo, Japan, 2Chiba University, Chiba, Japan. Connectin is the largest protein that connects between Z-line and M-line of sarcomere and functions as a molecular spring of vertebrate striated muscles. At the contiguous region of connectin gene on mammalian genomes, there are two genes for proteins that function remain unknown. One protein (about 75kDa) consists of a SEC14 domain and three spectrin repeats, and the other protein (about 150kDa) consists of six spectrin repeats and an immunogloburin-like domain. We performed the RT-PCR experiments and revealed that these two genes are expressed in striated muscles. The western blot tests using newly produced antibodies also indicated that these proteins existed in striated muscles. The immunofluorescence microscopic observation Tuesday, February 10, 2015 elucidated that the 75kDa protein are located on the membrane of muscle fiber but the 150 kDa protein are localized at the Z-line of the sarcomere. The localization of these proteins are also confirmed by the transfection of GFP-fusion proteins into cultured skeletal muscle cells. These insights indicate that novel proteins that transcribed from the contiguous genes of connectin gene exist in striated muscles. 2117-Pos Board B254 Skeletal Myosin Binding Protein-C Isoforms Modulate Actomyosin Contractility and are Regulated by Phosphorylation Amy Li1,2, Samantha Beck Previs2, Michael Previs2, Brian Lin3, Cristobal dos Remedios1, Roger Craig4, Sakthivel Sadayappan3, David Warshaw2. 1 Anatomy & Histology, University of Sydney, Sydney, Australia, 2 Department of Molecular Physiology & Biophysics, University of Vermont, Burlington, VT, USA, 3Department of Cell and Molecular Physiology, Loyola University Chicago, Maywood, IL, USA, 4Department of Cell and Developmental Biology, University of Massachusetts Medical School, Worcester, MA, USA. Myosin binding protein C (MyBP-C) is a thick filament-associated protein found in striated muscle and may regulate muscle contractility. Separate genes encode the fast and slow skeletal isoforms, and there are potential PKA phosphorylation sites in their functionally important N-terminal regions. Here, we compare mouse N-terminal fast (fC1C2) and slow (sC1C2) skeletal fragments containing the initial ~50 aa Pro/Ala-rich domain and the C1 and C2 Igdomains that are linked by the ~100 aa M-domain. Of the known slow skeletal splice variants, we chose a highly expressed variant lacking the N-terminal 34-59 residue insert. To define the potential mechanisms by which skeletal MyBP-Cs affect contractility and whether these effects are modulated by PKA phosphorylation, we assessed the Ca2þ-dependent motility of rabbit skeletal native thin filaments over a surface of rabbit psoas myosin in the presence of C1C2 fragments. While thin filaments were fully regulated, with no motion observed at pCa > 7 in the absence of fragments, the addition of either 0.50 mM fC1C2 or sC1C2 resulted in significant motility. This suggests that skeletal MyBP-C isoforms effectively sensitize the thin filament. Under fully activating conditions (pCa % 5), sC1C2 had little effect on motility whereas fC1C2 inhibited sliding velocities by nearly 50%. Thus, these fragments differ in their modulatory capacities with fC1C2 sensitizing the thin filament to Ca2þ and inhibiting maximal velocities, while the sC1C2 variant exhibits only a single mode of contractile modulation; i.e. thin filament sensitization. Interestingly, PKA phosphorylation in the Pro/Ala (sC1C2) and M-domains (sC1C2 and fC1C2), as confirmed by mass spectrometry, reduced both fragments’ Ca2þ sensitization of the thin filament. Thus, the function of MyBPC isoforms may be tuned to match the physiological demands of the muscle in which they are expressed. 2118-Pos Board B255 In vitro Reconstitution of Skeletal Muscle Contraction using Native Thin Filaments Augustine Cleetus, Khushboo Rastogi, Ravikrishnan Elangovan. Department of Biochemical Engg and Biotechnology, Indian Institute of Technology, Delhi, India, New Delhi, India. Intracellular Ca2þ concentration regulate the muscle contraction by changing the conformation of Tropomyosin molecule over Actin filament. In this work we have purified native thin filaments (NTF) from Chicken Pectoralis muscle and reconstituted in in vitromotility assay (IVMA) with skeletal muscle myosin II. NTF prepared were 1.6750.14 mm in length and showed good regulation under different Ca2þ concentrations. At pCa 4, more than 90% of NTF were continuously sliding and when solution changed to pCa 9 all the filaments were rigidly stuck to surface. Sliding velocity of NTF was VF¼5.6351.21 mm/s at 30 oC and 17% higher compared to unregulated actin filaments. Fraction of sliding filament showed a direct correlation with pCa of buffer and pCa50¼ 6.69 was obtained using Hills equation fit. However the sliding filament velocity was not affected under different pCa concentration. Under varying substrate [MgATP] conditions in IVMA we found kþATP is 4.35 0.56 x 106 M1s1 and k-ADP is 404.3352.7 s1 at 30 oC. These kinetic parameters are comparable to values obtained using unregulated actin filaments, suggesting the detachment kinetics of myosin II is not affected due to Ca2þ regulation. We studied the effect of myosin density on thin filaments activation in IVMA. Even with fully activated thin filament (pCa 4.0), decrease in myosin density leads to increase in fraction of stuck filaments. Decrease in fraction of motile filaments with myosin density in IVMA suggests there is minimum number of myosin head binding required to completely activate the NTF. Supported by Department of Science and Technology, Government of India. 421a 2119-Pos Board B256 Direct Troponin-Myosin Interaction Enhances ATPase Activity of Cardiac HMM Nazanin Bohlooli Ghashghaee1, King-Lun Li1, Wen-Ji Dong1,2. 1 The Gene and Linda Voiland School of Chemical Engineering and Bioengineering, Washington State univeristy, Pullman, WA, USA, 2The Department of Integrative Physiology and Neuroscience, Washington State univeristy, Pullman, WA, USA. Phenomenological evidence has long suggested that the strongly bound crossbridges exert a positive feedback on myofilament regulation, which increases the sensitivity of thin filament to Ca2þ and achieves full activation. This positive feedback mechanism is uniquely important to the beat-to-beat regulation of cardiac output. Despite its importance, the molecular mechanism remains elusive. The current wisdom implies that the positive feedback mechanism is mediated by specific movement of tropomyosin on actin surface caused by actin-myosin interaction. Since the Ca2þ-regulated interactions between cardiac thin filament and myosin can be considered as an allosteric system which has long-range coupling between distinct components of the system, multiple protein-protein interactions at the interface between thin and thick filament may provide alternative mechanism for the feedback effects on myofilament activation. In this study, we investigated the potential interaction between troponin and myosin, known as troponin-bridge, and how the interaction affects function of myosin and thin filament regulation. The direct interaction between myosin and cardiac troponin was monitored using a sedimentation assay. Different reconstituted troponin complexes were incubated with myosin followed by ultra-centrifugation to pull down the myosin and the bound proteins. Western blot was used to identify the troponin subunits that were bound to myosin. Our results suggest that both N-cTnI(1-129) and C-cTnT (T2) bind to myosin, suggesting a possible direct interaction between myosin and IT arm of troponin. ATPase assay showed that the HMM ATPase activity was significantly enhanced by the presence of troponin regardless of the presence of actin and/or tropomyosin. In addition, truncation of the C-domain (residues 129-212) of cTnI also increased the HMM ATPase activity. The results of our study indicate that troponin-bridge may be a crucial component of thin filament regulation and play an important role in actomyosin interaction and muscle function. 2120-Pos Board B257 The Regulation of Actomyosin ATPase in Cardiac Muscle by the N-Terminal Extension of Cardiac Troponin T Laura Gunther1, Hanzhong Feng2, Hongguang Wei2, Justin Raupp1, Jian-Ping Jin2, Takeshi Sakamoto1. 1 Physics and Astronomy, Wayne State University, Detroit, MI, USA, 2 Physiology, Wayne State University School of Medicine, Detroit, MI, USA. Cardiac troponin T (cTnT) and cardiac troponin I (cTnI) both have an aminoterminal variable region that plays a role in modifying the overall protein conformation and functions in the calcium-dependent regulation of cardiac muscle function. Previous studies have showed that in comparison with wild type controls, transgenic mouse hearts over-expressing cTnT lacking the N-terminal variable region (cTnT-ND) had an increased rate of relaxation. In contrast, transgenic mouse heart over-expressing cTnI lacking the N-terminal extension (cTnI-ND) had lower sensitivity to calcium activation of ATPase, resulting enhanced ventricular relaxation and Frank-Starling response. Recently, we demonstrated that the second order mant-dATP binding rate of cardiac myofibrils containing cTnI-ND was three-fold as fast as that of wild type myofibrils in low [Ca2þ]. The ADP dissociation rate of cTnI-ND myofibrils was positively dependent on calcium concentrations, while the wild type controls were not significantly affected. These results from native cardiac myofibrils under physiological conditions indicate that the N-terminal extension of cTnI plays a role in the calcium regulation of the kinetics of actomyosin ATPase. In the present study, we employed the same techniques in order to investigate which step(s) of the ATPase cycle is regulated by the N-terminal variable region of cTnT. mantATP binding, mant-ADP/ADP dissociation, and phosphate releasing rate of cTnT-ND myofibrils are examined and the results will provide novel information dissecting the function of the two troponin subunits and their posttranslational modifications. 2121-Pos Board B258 Pseudo-Acetylation of Actin Residues K326 and K328 Disrupts Drosophila Flight Performance and Muscle Structure William M. Schmidt, Meera Cozhimuttam Viswanathan, Anna C. Blice-Baum, D. Brian Foster, Anthony Cammarato. Cardiology, Johns Hopkins University, Baltimore, MD, USA. Striated muscle contraction is driven by cyclical interaction between myosincontaining thick and actin-containing thin filaments and is regulated by the 422a Tuesday, February 10, 2015 tropomyosin-troponin complex. In the absence of calcium, troponin constrains tropomyosin in a blocking position where it shields myosin binding sites on actin. Calcium binding to troponin enables azimuthal movement of tropomyosin, which exposes myosin binding sites, and relieves contractile inhibition. Previous models indicated that K326 and K328 on actin form electrostatic contacts with E181 of tropomyosin in an inhibitory state (Li, et al. 2011), and that K328 also contacts E286 of myosin S1 during contraction (Behrmann, et al. 2012). A recent proteomic study showed that K326 and K328 are acetylated in guinea pig cardiac thin filaments (Foster, et al. 2013). Since this posttranslational modification would negate the positively charged lysines, and potentially ablate vital ionic interactions between actin and tropomyosin or myosin, we predicted K326 and K328 acetylation would alter muscle performance. We tested this hypothesis in vivo by expressing K326Q, K328Q, or K326Q/K328Q acetyl-mimetic actin in Drosophila muscle. Polarized light microscopy of K328Q and K326Q/K328Q indirect flight muscle revealed a severe disturbance to fiber structure, and flight tests confirmed complete loss in flight ability relative to control. Flies harboring only the K326Q mutation, however, had similar fiber structure to control, but nonetheless displayed a significant decrease in flight performance. These results suggest that K326 and K328 play important roles in maintaining proper muscle function. We are currently investigating the effects of the pseudo-acetylated actin residues on Drosophila hearts. Overall, our findings highlight the utility of Drosophila as a model that permits efficient targeted design and assessment of molecular and tissuespecific responses to muscle protein modifications, in the physiological context of muscle. 2122-Pos Board B259 Studying Troponin within Regulated Actin at Single Molecule Resolution Christopher Solis-Ocampo, Maria E. Moutsoglou, Gi-Ho Kim, John M. Robinson. Chemistry & Biochemistry, South Dakota State University, Brookings, SD, USA. In cardiac muscle troponin (Tn) and tropomyosin (Tm) associate with filamentous actin (Ac) to provide Ca2þ-dependent regulation of contraction. In situ, Ca2þ activation is a highly cooperative process thought to be linked to cooperative structural interactions between Tn, Tm, and Ac. The integrity of the regulated actin assembly is a concern at the low concentrations needed to achieve single molecule resolution. Previous studies of filament stability were based on indirect methods performed and conducted under conditions of high ionic strength. Here, we characterized the the binding of Tn-Tm to Ac under near physiologic conditions by directly visualizing dye-labeled reconstituted regulated actin filaments (rAc). Tn, Tm, and Ac were labeled, respectively, with AF546, ATTO655, and phalloidin-AF488. Regulated actin was reconstituted at 1:1:1 stoichiometry of Tn:Tm:Ac7. From the co-localization of the fluorescence emission, we observe that Tn-Tm binds non-cooperatively (Hill coefficient approaches 1) to actin with a dissociation constant of 0.1 mM. At concentrations greater than 100 nM, Tn-Tm binds to Ac in a all-or-nothing fashion, where Ac either has Tn-Tm bound along its length or has no Tn-Tm bound at all. At 500 nM Tn-Tm, approximately 83% of Tn-Tm in solution is bound to Ac; bound Tn-Tm decorates Ac confluently; unbound Tn-Tm dissociate from each other. By reconstituting regulated actin with sparsely labeled Tn, we are able to study Ca2þ-dependent Tn switching within regulated actin at single molecule resolution. This is confirmed by FCS measurements that suggest that less than one dye molecule is present in the confocal at any given time. 2123-Pos Board B260 Observing the pCa-Force Relationship with a 3-Bead Laser Trap Assay Thomas J. Longyear1, Sam Walcott2, Edward P. Debold1. 1 University of Massachusetts, Amherst, MA, USA, 2UC Davis, Davis, CA, USA. Muscular force is highly dependent on the amount of free Caþþ in the myoplasm because actomyosin binding is carefully regulated by troponin (Tn) and tropomyosin (Tm). To gain molecular insight into the Caþþ dependence of force, we directly observed the effect of Caþþ on the force-generating capacity of a mini-ensemble of myosin (~6 heads) interacting with a single reconstituted thin filament (actinþTnþTm) in a three bead laser trap assay at 100uM ATP. Maximum force decreased in a Caþþ-dependent manner (6.75, 4.75, 3.25, and 1.85pN at pCa 5, 6, 6.5 and 7 respectively). Furthermore, the average forces were significantly different (p<0.05) as Caþþ levels were decreased from pCa 5 (mean force 5 S.E. ¼ 0.94 50.033, N ¼ 1253) to 6 (0.47 5 0.014, N ¼ 585), 6.5 (0.5350.014, N ¼ 841), and 7 (0.3850.004, N¼2012). Saturating Caþþ (pCa5) produced a broad distribution of forces, ranging from 0.2 pN (consistent with single actomyosin events) to 6.75 pN (consistent with multiple successive actomyosin attachments). However, the distribution became tighter at lower pCa levels. Indeed, at pCa5, approximately 15% of the events produced > 1.5pN where only 0.1% of the events achieved this force level at pCa7. In addition, the total percent of time the myosin molecules were strongly bound to the thin filament were 41, 21, 16, and 14% at pCa 5, 6, 6.5 and 7 respectively. Overall, these data provide unique insight into the molecular events that underlie the Caþþ-dependence of force generation in a regulated system. 2124-Pos Board B261 Energy Landscapes Reveal the Myopathic Effects of Tropomyosin Mutations Marek Orzechowski1, Gerrie P. Farman1, Jeffrey R. Moore1, Stefan Fischer2, William Lehman1. 1 Dept. Physiology & Biophysics, Boston University School Medicine, Boston, MA, USA, 2IWR, University of Heidelberg, Heidelberg, Germany. Striated muscle contraction is regulated by a complex interaction network connecting the effects of troponin, Ca2þ, and myosin-heads to the azimuthal positioning of tropomyosin along thin filaments. In relaxed muscle, tropomyosin sterically blocks crossbridge cycling on actin to inhibit contraction, while in stimulated muscle, tropomyosin repositions over actin, and crossbridge cycling and contraction result. Many missense mutations, located at the actintropomyosin interface, however, reset the regulatory switching mechanism either by weakening or strengthening residue-specific interactions, leading to hyper- or hypo-contractile pathologies. We have computed energy landscapes for the actin-tropomyosin interface and have quantified contributions of single amino acid residues to actin-tropomyosin binding. The information acquired can prove to be a useful diagnostic tool to assess effects of actin and tropomyosin mutations by relating the critical initial stages of disease development to alterations in thin filament stability and regulation. We find that the landscapes for mutant filaments associated with hyper-contractility, for example those linked to hypertrophic cardiomyopathy (HCM), skeletal muscle arthrogryposis and congenital fiber-type disproportion (CFTD), provide a simple picture. In these cases, most of the mutations examined are associated with a decrease in actin-tropomyosin interaction energy that will destabilize the blocked (relaxed)-state. Our measurements parallel previously noted enhanced Ca2þsensitivity conferred by these mutants. We show, in addition, that energy landscape computation, in combination with known actin-tropomyosin sequence and structural information, can be used prospectively to identify potential effects of post-translational modifications to rescue regulatory imbalances. However, unlike results on hyper-contractility-related mutants, landscapes for tropomyosin mutants tied to hypo-contractility do not present a straightforward picture. These mutations may affect other components of the regulatory network, e.g., troponin-tropomyosin signaling or tropomyosin-myosin interaction. 2125-Pos Board B262 Estimation of Local Forces in Myofilaments using X-Ray Diffraction Patterns and Muscle Mechanics Data Momcilo Prodanovic1, Djordje Nedic2, Thomas C. Irving1, Srboljub M. Mijailovich3. 1 BCPS, Illinois Institute of Technology, Chicago, IL, USA, 2University of Kragujevac, Kragujevac, Serbia, 3Northeastern University, Boston, MA, USA. Small-angle X-ray diffraction of contracting muscle provide a view of sarcomere structure averaged over the area illuminated by the X-ray beam. Many important details of muscle contraction, however, such as crossbridge attachment and detachment rates are likely to depend on the local structures and forces on these structures. We have developed a methodology for predicting X-ray diffraction patterns with stepwise increases of strain along actin filaments in the hexagonal sarcomere lattice. Using PDB data for the crystal structures of G-actin we reconstructed the geometry of actin filaments deformed under stress generated by crossbridges. This stress varies along the filament length, so these deformations also vary with length. These fiber deformations may be determined by Monte Carlo calculations using the computational platform, MUSICO, along with the force and length changes seen during classical mechanical protocols. Using the predicted changes in spacings of actin filaments due to crossbridge forces we predicted how the X-ray diffraction patterns vary along with force and length during mechanical transients. Predicted X-ray diffraction patterns of fully developed isometric force muscle fibers are compared to experimental X-ray diffraction measurements in order to estimate the average force in actin filaments. Using peak shape analysis of the meridional reflections, one can assess the variation of filament deformation, and hence force, along the length of the filaments. Using the same methodology, we estimated the local modulation of force on actin during different mechanical protocols, including Huxley-Simmons quick releases and isotonic shortening. These data enable us to better correlate the molecular events, such as the current Tuesday, February 10, 2015 number of attached crossbridges and the distributions of crossbridge forces to macroscopic measurements of force and length changes. Supported by: NIH 9 P41 GM103622, R01 AR048776 and R01 DC 011528 2126-Pos Board B263 Hierarchy of Regulatory Interactions in the Sarcoplasmic Reticulum Calcium Transport Complex John E. Rubin1, Bengt Svensson1, Kurt C. Peterson1, Seth L. Robia2, David D. Thomas1, Joseph M. Autry1. 1 Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN, USA, 2Cell and Molecular Physiology, Loyola University, Maywood, IL, USA. We have used fluorescence microscopy to detect interactions of sarcolipin (SLN), phospholamban (PLB), and the SR Ca-ATPase (SERCA1a isoform). SLN and PLB individually regulate SERCA activity through protein-protein interactions controlled by phosphorylation; SLN is highly expressed in fasttwitch muscle and atria, while PLB is highly expressed in slow-twitch muscle and ventricles. When SERCA, SLN, and PLB are expressed in the same human cell (vastus lateralis muscle, Takotsubo cardiomyopathy), the three proteins form a ‘‘super-inhibited’’ ternary complex (SERCA-SLN-PLB), whereby SERCA activity shows 5-fold decreased calcium affinity and 2-fold decreased maximum velocity (MacLennan JBC 2002; Tupling PLOS 2013). Here we used Fo¨rster resonance energy transfer (FRET) to quantify the complex equilibria of homo- and hetero-oligomeric interactions between SERCA, SLN, and PLB. The three proteins were tagged with genetically-encoded fluorescent probes (CFP, YFP), and the fluorescent fusion proteins were expressed in Sf21 cells via baculovirus infection. Five protein-protein interactions were assayed (SLN-SLN, SLN-PLB, PLB-PLB, SERCA-SLN, SERCA-PLB), and three parameters were calculated per interaction (binding affinity, oligomer number, interprobe distance). Results indicate (a) SLN and PLB show high-affinity selfassociation into homo-oligomers and low-affinity cross-assembly as heterodimers, and (b) SERCA forms 1:1 binary complexes with SLN or PLB when co-expressed with either subunit individually, with SERCA having 3-fold higher affinity for SLN over PLB. We conclude that SLN and PLB monomers bind independently to SERCA, and that each subunit shows competing selfassociation versus regulatory complex formation. Molecular modeling based on FRET parameters was used to examine the SERCA-SLN-PLB complex. We propose that equilibrium allocation of SERCA between binary and ternary complexes depends on the expression level and phosphorylation state of each regulatory subunit in muscle. Acknowledgments: This work was funded by NIH grants to DDT (GM27906, AR0507220, AR007612) and UMN awards to JMA (CBS-HHMI). 2127-Pos Board B264 Regulation of Myoblast Proliferation and Differentiation by Anoctamin 5 and 6 Li Xu, Renzhi Han, LiXia Zhao. Ohio State University, Columbus, OH, USA. Anoctamin 5 (ANO5) and 6 (ANO6) belong to a conserved gene family (anoctamins, also known as TMEM16 ), which encode for proteins predicted to have eight transmembrane domains and putative Ca2þ-activated chloride channel (CaCC) activity. Genetic defects in this gene family result in a number of human diseases. In particular, mutations in ANO5 have been linked to gnathodiaphyseal dysplasia 1 (GDD1), limb-girdle muscular dystrophy (LGMD2L), and Miyoshi myopathy (MMD3), while mutations in ANO6 cause a bleeding syndrome. However, the biological functions of these anoctamins in skeletal muscle are largely unknown. In this study we examined the roles of Ano5 and Ano6 in C2C12 proliferation and differentiation. Our data showed that Ano6 but not Ano5 plays an essential role in C2C12 myoblast proliferation, likely via regulating the ERK/AKT signaling pathway. In addition, Ano5 or Ano6deficiency alone does not affect the myogenic differentiation program of C2C12 cells, however knocking down both Ano5and Ano6 remarkably promotes C2C12 myoblast differentiation. Our data demonstrate that Ano5 and Ano6 together may play an important role in regulating the switch from proliferation to differentiation during myogenesis. Further investigation will focus on the mechanisms of Ano5 and Ano6 in this regulatory process. 2128-Pos Board B265 Charged Vesicles Potently Induce NLRP3 Inflammasome Activation Lixia Zhao1, Li Xu1, Zhenyu Zhong2, Yougang Zhai2, Liang Qiao2, Renzhi Han1. 1 The OHIO STATE UNIVERSITY, Columbus, OH, USA, 2Loyola University Chicago, Maywood, IL, USA. Extensive muscle inflammation is frequently observed in muscular dystrophy patients, particularly in those with dysferlin deficiency. Dysferlin has been 423a shown to play multiple functions in skeletal muscle including the plasma membrane repair. However, the mechanisms underlying the extensive muscle inflammation in dysferlin-deficient muscular dystrophy are largely unknown. Our previous study revealed that genetic ablation of the complement system ameliorates the pathology in a mouse model of dysferlinopathy, indicating that the innate immune activation may be involved in the pathogenesis. However, it is unclear how the innate immune system contributes to the disease pathology in dysferlinopathy. Recently, we found that vesicles composed of charged lipids can potently activate the NLRP3 inflammasome, resulting in the maturation and release of pro-inflammatory cytokine interleukin-1b. Furthermore, mitochondrial reactive oxygen species-dependent calcium influx via the TRPM2 channel plays an essential role in activation of the NLRP3 inflammasome. Interestingly, NLRP3 inflammasome pathway has been shown to be up regulated in dysferlin-deficient muscle. To further investigate the regulatory signaling pathways involved in NLRP3 inflammasome activation, we established a cell screening system using THP-1 cells. NLRP3 inflammasome activation induces pyroptosis, a form of programmed cell death associated with antimicrobial responses during inflammation. LPS was used to stimulate PMA-differentiated THP1 cells, Nigericin was used to induce NLRP3 activation and over 80% cell death. To validate the system, we created NLRP3-deficient THP-1 cells using CRISPR technology. The NLPR3deficient THP-1 cells showed significantly decreased cell death when compared to control cells. We will use this system to identify novel regulators of NLRP3 inflammasome. 2129-Pos Board B266 Mitsugumin 56 is an MBOAT Family Member and Contributes to Postnatal Muscle Maturation Myuki Nishi1, Bo Van1, Shinji Komazaki2, Daiju Yamazaki1, Ki-Ho Park3, Jianjie Ma3, Hiroshi Takeshima1. 1 Biological Chemistry, Kyoto University Graduate school of Pharmaceutical Sciences, Kyoto, Japan, 2Anatomy, Saitama Medical University, Saitama, Japan, 3Surgery, The Ohio State University, Columbus, OH, USA. The sarcoplasmic reticulum (SR) functions as a highly specialized intracellular Ca2þ store controlling muscle contraction cycle. Although major SR proteins involved in Ca2þ handling have been extensively studied, there are still SR components with unknown functions, which would potentially open new study fields in muscle biology. Here we report mitsugumin 56 (MG56), a new SR membrane protein predominantly expressed in striated muscle. MG56 belongs to the MBOAT (membrane-bound O-acyltransferase) family, and is specifically localized to the junctional SR composing the triad in skeletal muscle. Mg56-knockout mice grew normally for a week after birth, however, they gradually developed suckling failure and died within two weeks under starvation conditions. In the skeletal muscle of Mg56-knockout mice, the SR elements began to swell near the Z-line prior to physical debilitation, and further developed enormous vacuoles spreading over the sarcomeres. However, in tension measurements, regular contractile features were largely preserved in Mg56knockout muscle that contained swelling SR elements. Meanwhile, biochemical analysis demonstrated that unfolded protein response was highly activated in Mg56-knockout muscle, suggesting that the suckling failure was caused by disrupted muscle maturation under ER stress conditions. Therefore, MG56 exerts anti-ER stress activity in the developing SR and is essential for postnatal muscle maturation. 2130-Pos Board B267 Suppressed Autophagy Flux in Skeletal Muscle of an Amyotrophic Lateral Sclerosis Mouse Model Yajuan Xiao1,2, Changling Ma1, Jianxun Yi1,2, Shaoping Wu1, Guo Luo1, Xiulong Xu1, Pei-Hui Lin3, Jun Sun1, Jingsong Zhou1,2. 1 Rush University, Chicago, IL, USA, 2Kansas City University of Medicine and Biosciences, Kansas City, MO, USA, 3Davis Heart and Lung Research Institute, The Ohio University Wexner Medical Center, Columbus, OH, USA. Amyotrophic lateral sclerosis (ALS) is a fatal neuromuscular disease characterized by the progressive loss of motor neuron and skeletal muscle atrophy. Accumulation of abnormal protein inclusions is implicated in motor neuron degeneration in ALS. Autophagy, an intracellular process targeting misfolded proteins and damaged organelles for lysosomal degradation, plays crucial roles in survival and diseased conditions. Efforts were made to understand the role of autophagy in motor neuron degeneration and to target autophagy in motor neuron for ALS treatment. However, results were quite contradictory. Skeletal muscle comprises around 40% of whole-body lean mass and is substantially affected in ALS. Possible autophagy defects in skeletal muscle may also complicate the results. Here, we examined autophagy activity in skeletal muscle of an ALS mouse model G93A. Through overexpression of a fluorescent protein LC3-RFP, we found a basal increase in autophagosome formation in 424a Tuesday, February 10, 2015 G93A muscle during disease progression when the mice were on a regular diet. As expected, an autophagy induction procedure (starvation plus colchicine) enhanced autophagy flux in skeletal muscle of normal mice. However, in response to the same autophagy induction procedure, G93A muscle showed significant reduction in the autophagy flux. Immunoblot analysis revealed that increased cleaved caspase-3 associated with apoptosis was linked to the cleavage of several key proteins involved in autophagy, including Beclin-1, which is an essential molecule connecting autophagy and apoptosis pathways. Our data suggest that the cytoprotective autophagy pathway is suppressed in G93A skeletal muscle and this suppression may link to the enhanced apoptosis during ALS progression. The abnormal autophagy activity in skeletal muscle likely contributes muscle degeneration and disease progression in ALS. 2131-Pos Board B268 Impairment in Acetylcholine Release by Cardiomyocytes Leads to Enhanced Pathological Hypertrophy Cibele Rocha-Resende1, Vania Prado2, Marco Prado2, Aristobolo Mendes Silva3, Silvia Guatimosim1. 1 Physiology, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil, 2 Robarts Research Institute, London, ON, Canada, 3Morphology, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil. Our group has previously demonstrated that cardiomyocytes express ChAT (choline acetyltransferase), VAChT (vesicular acetylcholine transporter) and ChT-1 (choline high affinity transporter), proteins involved in the synthesis, release and reuptake of acetylcholine. Additionally, we have shown that this cardiomyocyte intrinsic cholinergic machinery has an important role in preventing deleterious effects of adrenergic signaling in vitro and in vivo. To address the role of this non-neuronal acetylcholine for heart function, a new mouse lineage was generated with deletion of VAChT (vesicular acetylcholine transporter) only in cardiac myocytes (cVAChT-ko). These mice are characterized by heart hypertrophy, calcium signaling dysfunction and increased reactive oxygen species levels. In order to better understand how impairment in acetylcholine released from cardiomyocytes could impact heart function, we subjected these mice to transverse aortic constriction (TAC) and evaluated left ventricular hypertrophy at 3 and 14 days after surgery. 3 days after TAC, cardiac hypertrophy was more prominent in cVAChT-ko mice than in control hearts (control: 11% versus cVAChT: 24%). However, 14 days after surgery, heart and left ventricular hypertrophy were similar between cVAChT-ko and control mice. Additionally, the lung/body ratio was significantly increased only in cVAChT-ko with constriction (control: 9% versus cVAChT-ko: 54%), suggesting that these mice present pulmonary edema, which is an indicative of heart failure. Moreover, TAC induced an increase in PLN/SERCA ratio only in cVAChT-ko mice. Thus our data show that cVAChT-ko mice is more prone to cardiac disease under stress conditions than control mice, indicating that cardiomyocyte intrinsic cholinergic machinery plays an important role in cardiac protection. FINANCIAL SUPPORT: CNPq, FAPEMIG, NIH (Fogarty International Award) 2132-Pos Board B269 Myopathic Changes in Murine Skeletal Muscle Lacking Synemin ´ Neill1, Karla Garcia-Pelagio1, Joaquin Muriel1, Andrea O Patrick Desmond1, Richard M. Lovering1, Linda Lund2, Meredith Bond3, Robert Bloch1. 1 University of Maryland, Baltimore, MD, USA, 2University of Baltimore, Baltimore, MD, USA, 3Cleveland State University, Cleveland, OH, USA. Diseases of striated muscle linked to intermediate filament (IF) proteins are associated with defects in the organization of the contractile apparatus and its links to costameres, which connect the sarcomeres to the cell membrane. Here we study the role in skeletal muscle of synemin, a type IV IF protein, by examining mice null for synemin (synm-null). Synm-null mice have a mild skeletal muscle phenotype. Tibialis anterior muscles show a significant decrease in mean fiber diameter, in twitch and tetanic tension and an increase in susceptibility to injury caused by lengthening contractions. Organization of proteins associated with the contractile apparatus and costameres is not significantly altered in the synm-null. Elastimetry of the sarcolemma and associated contractile apparatus in extensor digitorum longus myofibers reveals a reduction in stiffness consistent with an increase in sarcolemmal deformability. Although fatigue with repeated isometric contractions is greater in isolated TA muscles of synm-null mice, the ability of the mice to run uphill on a treadmill is similar to controls. Our results show that synemin contributes to linkage between costameres and the contractile apparatus, and that the absence of synemin results in decreased fiber size and increased sarcolemmal deformability and susceptibility to injury. Synemin plays a moderate but distinct role in fast twitch skeletal muscle. Supported partially by a Physiological Genomics and a CONACyT fellowship to KPGP, and by NIH to RML (R01 AR 059179), to MB (RO1 02520711), and to RJB (R01 AR 055928). 2133-Pos Board B270 Cytokine Stimulation Induces Nox2-Dependent ROS Production and Decreases Muscle Function James A. Loehr, Reem Abo-Zahrah, Rituraj Pal, George G. Rodney. Baylor College of Medicine, Houston, TX, USA. Increased reactive oxygen species (ROS) are a hallmark of many diseases, such as inflammatory myopathies. Recent evidence suggests elevated cytokine activity increases ROS production resulting in muscle weakness; however, the specific source of ROS production has yet to be fully elucidated. Redox sensitive probes, targeted to NADPH oxidase 2 (Nox2) (p47-roGFP) and the mitochondria (mito-roGFP), were used to assess the sub-cellular site of ROS production in the presence of various cytokines. In addition, we assessed the effect of cytokine induced ROS production on skeletal muscle function. Cytokine stimulation increased p47-roGFP oxidation approximately 15%, but had no effect on mito-roGFP oxidation. Genetic and pharmacological inhibition of Nox2 resulted in decreased Nox2-dependent ROS production while genetic overexpression of SOD2 had no effect on mitochondrial or Nox2-mediated ROS production. Following cytokine administration, skeletal muscle function decreased by 30% and genetic inhibition of Nox2-activity partially rescued muscle function. Genetic inhibition of mitochondrial-ROS provided no protection against decreased muscle function following cytokine stimulation. Collectively, these data indicate that elevated cytokine activity resulted in increased ROS production at specific sub-cellular sites, negatively affecting muscle function. Our data highlight the importance of understanding the source of ROS production in response to physiological and/or pathological stimuli such that targeted therapeutic approaches can be developed to combat the deleterious effects of oxidative stress. Mechanisms of Voltage Sensing and Gating 2134-Pos Board B271 An Experimentally-Validated Model Structure of the Hv1 Proton Channel Voltage Sensor in its Resting State Younes Mokrab1, Ashley Bennett2, Mark S.P. Sansom3, I. Scott Ramsey2. 1 Neuroscience Research and Development, Eli Lilly and Company, Windlesham, United Kingdom, 2Dept Physio/Biophys, Virginia Commonwealth Univ, Richmond, VA, USA, 3Bipchemistry, Oxford University, Oxford, United Kingdom. The structure of an Hv1-based chimeric voltage sensor protein (mHv1cc) was recently solved by X-ray crystallography. Although mHv1cc mediates voltagegated Hþ currents when expressed in mammalian cells, the crystallized protein likely represents a closed, possibly resting, conformation. However, the mHv1cc structure is apparently incompatible with our experimental measurements of the resting-state Hþ ‘shuttle’ conductance (GSH) in hHv1 R205H. In order to explore possible conformations of the Hv1 voltage sensor (VS) domain in its resting state, we created an Hv1 VS domain homology model (Hv1 D) based on a previously reported resting-state Rosetta model structure of the Shaker Kþ channel VS domain (Pathak, et al. 2007) and subjected the Hv1 model to all-atom MD simulations. We subsequently created a model of the Hv1 D R205H point mutant that mediates GSH. In contrast to mHv1cc and a resting-state model of the Ciona Hv1 VS domain (Chamberlin, et al. 2013), the central crevice in our new model is not obviously occluded by hydrophobic side chains. A prominent feature of Hv1 D R205H is that the imidazole ring of the introduced histidine appears to be simultaneously accessible to intra- and extra-cellular aqueous vestiblues, and therefore appears competent for Grotthuss-type proton conduction. Hv1 D represents the first VS domain resting-state model structure that satisfies the rigorous structural constraints imposed by experimental data and thus serves as a template for understanding the structural basis of VS activation in a variety of VS domain-containing proteins. 2135-Pos Board B272 Unveiling Potential Binding Sites in the Hv1 Four Helix Bundle Eleonora Gianti1, Lucie Delemotte1, Vincenzo Carnevale1, Francesco Tombola2, Douglas Tobias2, Michael L. Klein1. 1 Chemistry and the ICMS, Temple University, Philadelphia, PA, USA, 2 University of California, Irvine, CA, USA. Hv1 is a voltage-sensor protein that plays essential roles in proton conduction, pH homeostasis and production of high-level superoxide by phagocytes. Unregulated Hv1 activity has pathological implications, such as hyperproliferation of cancer cells and exacerbation of brain damage in ischemic stroke. Tuesday, February 10, 2015 Unlike other voltage-gated ion channels, Hv1 is composed by two subunits each containing a voltage-sensing domain (VSD) and lacking the pore domain. On the other hand, Hv1 shares striking similarities with the M2 proton channel of the influenza A virus (A/M2): in both cases a bundle of four helices lines a proton conduction pathway and positively charged ‘‘fragment-like’’ molecules act as blockers. Starting from the recently solved structure of Hv1, we combined homology modeling, molecular dynamics simulations and site finding methods to unveil potential binding pockets in the most relevant conformational states. We discovered that, similarly to the case of A/M2, the distribution of water molecules inside the channel lumen is state-dependent, offering a rationale to designing novel pore blockers. 2136-Pos Board B273 Allosteric Coupling between Open Subunits in the Hv1 Proton Channel Probed by Guanidino-Thiazoles Liang Hong1, Vikrant Singh2, Heike Wulff2, Francesco Tombola1. 1 Department of Physiology and Biophysics, UC Irvine, Irvine, CA, USA, 2 Department of Pharmacology, UC Davis, Davis, CA, USA. The Hv1 channel extrudes protons from cells in response to membrane depolarization, and regulates the production of reactive oxygen species by NOX enzymes. Its excessive activity has been implicated in cancer development and in brain damage during ischemic stroke. The channel is a dimeric complex made of two voltage-sensing domains (VSDs), each containing a gated proton permeation pathway. We have previously identified 2-guanidinobenzymidazole (2GBI) derivatives that block proton permeation through the two VSDs (Hong et al. 2013, Neuron 77(2): 274-286). For these compounds, the binding occurs independently in the two open subunits, and resembles the binding to a monomeric version of the channel. Here we describe a separate class of guanidine derivatives - 2-guanidinothiazoles - that block Hv1 in a cooperative way. Comparison of the dose dependences of inhibition of dimeric and monomeric channels shows that blocker binding to one open subunit causes an increase in binding affinity of the neighboring open subunit. Using a site-directed mutagenesis approach, we identify the residues in the binding site responsible for cooperativity and then explore the residues at the interface between subunits that mediate allosteric coupling in the open state. Understanding the molecular features of the inhibitor that modulate cooperative binding can help develop better drugs targeting the Hv1 channel. This work is supported by NIH -National Institute of General Medical Sciences, grant R01GM098973. 2137-Pos Board B274 Characterization and Subcellular Localization of Hv1 in Lingulodinium Polyedrum Confirms its Role in Bioluminescence Juan D. Rodriguez1, Saddef Haq2, Kristine F. Nowak1, Deri Morgan3, Vladimir V. Cherny3, Steven Bernstein1, Maredith S. Sapp1, John R. Curcuru1, Coretha Antchouey1, Scott J. Nowak1, Allen Place2, Thomas E. DeCoursey3, Susan M.E. Smith1. 1 Kennesaw State University, Kennesaw, GA, USA, 2University of Maryland Center for Environmental Sciences Columbus Center, Baltimore, MD, USA, 3 Department of Molecular Biophysics and Physiology, Rush University Medical Center, Chicago, IL, USA. In 1972, J. Woodland Hastings and colleagues predicted the existence of a proton selective channel that opens in response to depolarizing voltage (HV1) across the vacuole membrane of bioluminescent dinoflagellates and conducts protons into specialized luminescence compartments (scintillons), thus causing the pH drop that triggers the light flash. RNA-Seq data from several luminescent dinoflagellate species provided candidate HV1 genes. When expressed in mammalian cells, the predicted HV1 from Lingulodinium polyedrum displays the hallmark properties of bona fide proton channels, including time-dependent opening with depolarization, perfect proton selectivity, and characteristic DpH dependent gating. RT-PCR and Western blotting confirm expression of HV1 in L. polyedrum. Fluorescence confocal microscopy of L. polyedrum cells stained with antibodies to luminescence proteins luciferase and luciferin binding protein and to HV1 reveal structures consistent with HV1’s proposed function in bioluminescence. [Supported by NSF: MCB-1242985, NIH: GM102336, /] 2138-Pos Board B275 Engineered Voltage Sensing Phosphatases: What do they tell us about the Gating Mechanism? Angeliki Mavrantoni, Kirstin Hobiger, Michael G. Leitner, Dominik Oliver, Christian R. Halaszovich. Neurophysiology, Philipps-University Marburg, Marburg, Germany. Voltage sensing phosphatases (VSPs) contain a phosphoinositide phosphatase domain (PD), which is under the control of a voltage sensor domain (VSD). 425a Here, the underlying coupling mechanism is not fully understood. Recently, Liu et al. (NSMB 2012) and Hobiger et al. (Biophys J 2012, PLoS one 2013) proposed for Ciona intestinalis (Ci-) VSP an interaction of positively charged amino acid residues in the linker, which connects the PD to the VSD, with the negatively charged residue Asp400 in the ‘TI’ loop of the PD. In light of these findings we revisited the engineered VSPs PTENCiV (Lacroix et al., JBC 2011) and hVSP1CiV (Halaszovich et al., J Lipid Res 2012), chimeras consisting of Ci-VSP’s VSD and the PD of PTEN or hVSP1, respectively. In those chimeras Asp400 (Ci-VSP numbering) is not conserved. Nonetheless, they show robust voltage dependent phosphatase activity. Mutations within the linker showed effects mirroring those previously reported for CiVSP, suggesting a gating mechanism similar to the one found in wild-type Ci-VSP. We mutated Asp400 in Ci-VSP to Asn or Arg and the corresponding Asn (hVSP1) or Arg (PTEN) in the chimeric enzymes to Asp and measured their voltage dependent enzymatic activity as well as sensing currents to elucidate the importance of Asp400 for VSD-PD-coupling. We propose that the interactions involving Asp400 described by Liu et al. and Hobiger et al. are not a necessary prerequisite for the voltage dependent activation of VSPs, but allow for a more efficient VSD-to-PD coupling. Further work will be required to establish the essential interactions between VSP, linker, and PD. Supported by Deutsche Forschungsgemeinschaft (DFG, grant SFB 593, TP12 to D.O.) and University Medical Center Giessen and Marburg (UKGM, grant 32/2011MR to C.R.H.) 2139-Pos Board B276 The Role of the C2 Domain of Voltage Sensing Phosphatase (VSP) Kevin D. Zolman, Paul M. Castle, Susy C. Kohout. Cell Biology and Neuroscience, Montana State University, Bozeman, MT, USA. The voltage-sensing phosphatase (VSP) is a voltage-activated enzyme that dephosphorylates phosphatidylinositol phosphates (PIPs). VSP is unique because it is the only example of an enzyme activated by voltage, providing the first direct link between lipid signaling pathways and membrane potential. Along with the voltage sensing domain, VSP also has a catalytic domain and a C2 domain. To date, the role of the VSP C2 domain is not understood. C2 domains are generally known as lipid binding domains, however, previous work has suggested the VSP C2 has a role in catalytic activity. Specifically, the crystal structures [1,2] show the 522-loop of the C2 forms a portion of the enzyme active site and the mutation Y522A altered enzyme activity [2]. We further probed the role of Y522 by introducing a phenylalanine instead of an alanine. We found the Y522F mutation also shifts the voltage dependence of catalytic activity, suggesting that hydrogen bonding is not a factor when this residue participates in VSP activity. A role in catalysis does not exclude the VSP C2 from also contributing to lipid binding. To investigate this possibility, we started by fully deleting the C2 domain from Ci-VSP and found that the presence of the full C2 domain is necessary for normal phosphatase function. We then combined more specific C2 mutations with the ability to manipulate PIP concentrations using a rapamycin-induced system in order to further address the possible roles of the C2 in VSP function. Our goal is to investigate how the C2 domain contributes to VSP function. [1] Matsuda, M. et al. JBC, 286, 23368-77 (2011). [2] Liu, L. et al. NSMB, 19, 633-641 (2012). 2140-Pos Board B277 Investigating the Function of a Novel Voltage-Sensing Protein Erika Babikow, Ferenc Papp, Suvendu Lomash, Jamie Smith, Kenton Swartz. NIH, Bethesda, MD, USA. We have identified a protein coded by the C15orf27 gene that we named NVS (Novel Voltage Sensor). NVS contains 531 residues, and contains an S1-S4 domain, a 90 residue N-terminus and a 307 residue C-terminus, both of which are predicted to be intracellular. The most critical residues found in S1-S4 domains of other voltage sensors are conserved in NVS, including 3 Arg and a Lys in the S4 helix, 4 conserved acidic residues in S1-S3 and the charge-transfer Phe in S2. In addition, the C-terminus is predicted to contain a coiled-coil domain, similar to voltage-activated proton (Hv1) channels. Our hypothesis is that NVS functions as a voltage sensor that couples to intracellular signaling pathways or interacts with Hv1 to form heteroligomers through the C-terminal coiled-coil domain. We used site-specific voltage-clamp fluorometry and identified several positions at the outer ends of S3 and S4 where labeled Cys residues produced changes in fluorescence as a function of membrane potential. Several positions give complex fluorescence responses, starting with a rapid increase in fluorescence followed by slower decrease in fluorescence. We also investigated whether NVS can oligomerize with Hv1, but observe no change 426a Tuesday, February 10, 2015 in the gating properties of Hv1 when coexpressed with NVS, and NVS was not capable of interacting with Hv1 in pull down assays. Having no apparent interaction with Hv1, we set out to determine whether NVS forms oligomers. Using chemical crosslinking and pull-down assays, we see formation of oligomers that are consistent with dimers and dependent on the C-terminus. Taken together, our results support the hypothesis that NVS is a voltage sensing protein capable of dimerization, with a function independent of Hv1. We are currently using these findings and approaches to investigate the structural assembly of NVS and to identify interaction partners. 2141-Pos Board B278 Putative Voltage Sensitive Enzymes in Prokaryotes Joshua P. Clark, Susan M.E. Smith. Biology, Kennesaw State University, Kennesaw, GA, USA. A voltage sensor domain (VSD) is a protein module that rearranges its conformation based on the electric potential of the cell membrane. VSDs have classically been described as N-terminal modules that confer voltage sensitivity to C-terminal pore domains in ion channels. More recently, N-terminal VSDs have been shown to confer voltage response to C-terminal enzyme modules in voltage sensitive enzymes (VSE), while the isolated VSDs of voltage gated proton channels (Hv1) perform both voltage sensing and proton channel functions. So far, VSEs and Hv1s have been found only in eukaryotes. We have identified a set of prokaryotic sequences that contain a VSD homolog; however, the C-terminal domains of these sequences, which we refer to as putative prokaryotic voltage sensitive enzymes (ppVSE), are dramatically different from ion pores. As expected, predicted secondary structures of the N-terminal domain are similar to those for bona fide VSDs; however, unlike the pore domains of ion channels, which contain two transmembrane helices, predicted structures of the C-terminal domains of the ppVSEs do not contain transmembrane helices. Alignment of individual domains to the HMM of the ion channel pfam pf00520 indicates significant similarity of ppVSE N-terminal domains but no detectable similarity of ppVSE C-terminal domains to the pf00520 HMM. A phylogenetic analysis of VSDs from prokaryotic sequences indicates a distinct lineage of the ppVSE VSD. This is the first documented evidence of a prokaryotic VSD-containing protein that does not have a pore domain. 2142-Pos Board B279 Sequence Signature of Voltage Sensing Detected via Dimensionality Reduction Techniques Daniele Granata1, Matteo Marsili2, Michael L. Klein1, Vincenzo Carnevale1. 1 Institute for Computational and Molecular Science, Temple University, Philadelphia, PA, USA, 2Quantitative Biology, ICTP, Trieste, Italy. The family of six-transmembrane-helices channels shows recognizable sequence homology and a strictly conserved structural architecture; yet these channels are involved in a significantly heterogeneous set of physiological functions, ranging from reporting noxious environmental conditions, to shaping the neuronal action potential, to syncing the beating of the heart. A striking example of this heterogeneity is provided by the comparison between the polymodal transient receptor channels (TRPs), whose activation can be regulated by several stimuli, and the voltage-gated ion channels (VGCs), which are activated by the variations of the transmembrane potential through. By analyzing multiple sequence alignments spanning the first four transmembrane segments (the voltage sensor domain in VGCs) and comparing TRPs and VGCs, we highlight the sequence determinants of voltage-driven activation. To this end we exploit the concept of fractal dimension to characterize the complexity of the two datasets. Moreover, we use a novel feature-selection approach, to identify the socalled maximally informative samples, i.e. set of residues whose distribution is maximally informative about the selection pressure that generated and differentiate the sequence ensembles. 2143-Pos Board B280 Lipid-Dependent Conformational Transitions in KvAP are Driven by Voltage Sensing Domain Qufei Li, Julia Skalska, Sherry Wanderling, Eduardo Perozo. Biochemisty & Molecular Biology, University of Chicago, Chicago, IL, USA. Recent electrophysiological studies have shown that the mechanism of voltagedriven gating in Kv channels is exquisitely sensitive to the composition of the lipid membrane. For instance, non-phosphate lipids can dramatically right-shift the voltage dependence (G-V curve) of KvAP (1,2) in a way that promotes the stabilization of the ‘‘down’’ conformation of the voltage sensor. We evaluated the conformations of KvAP’s isolated VSD by means of site directed spin labeling CW EPR and DEER spectroscopy through reconstitution in lipids with (POPC:POPG) or without (DOTAP) phosphate groups. Our data suggested a novel Tilt-Shift model for the mechanism of voltage sensing with a ˚ upward tilt and simultaneous ~2 A ˚ axial shift of S4 (3). We have extended ~3 A these measurements to evaluate the effects on the KvAP pore domain and quantify the DOTAP molar fraction dependence on the conformation of the inner gate. While in the full-length channel DOTAP reconstitution triggers conformational rearrangements in both the S4 segment and the S6 inner bundle gate, in the absence of voltage sensing domain, DOTAP is unable to generate significant rearrangements in S6. This result is consistent with the idea that S4 movements are allosterically transmitted to the inner bundle gate. Although the mechanistic correlation between voltage-dependent and lipid-dependent gating in voltage sensing domains remains to be fully characterized, it is clear that just as with electric fields, the interacting lipids play a determinant role in defining the equilibrium between the activated and resting states of voltage sensing domain. 1. D. Schmidt, Q. X. Jiang, R. MacKinnon, Nature 444, 775 (2006) 2. H. Zheng, W. Liu, L. Y. Anderson, Q. X. Jiang, Nat Commun 2, 250 (2011) 3. Q. Li, S. Wanderling, P. Sompornpisut, E. Perozo, Nat Mol Struct Bio l21, 244 (2014) 2144-Pos Board B281 Molecular Determinants of Temperature Dependent Gating of Ion Channels Sandipan Chowdhury1, Brian W. Jarecki2, Baron Chanda3. 1 Vollum Institute, Oregon Health and Science University, Portland, OR, USA, 2Cellular Dynamics International, Madison, WI, USA, 3Neuroscience, University of Wisconsin Madison, Madison, WI, USA. Physiological sensation of heat or cold in higher organisms is mediated by specialized ion channels whose opening and closing is exquisitely regulated by ambient temperatures. Members of TRP channel family, a branch of the much larger voltage-gated ion channel superfamily, serve as the primary physiological thermo-sensors. However, the physicochemical underpinnings of high temperature-sensitivity of channel gating remain poorly understood. Here, using a heuristic protein design approach, we have transmuted a temperatureinsensitive potassium channel into a heat or a cold-sensitive channel. By varying amino acid polarities at sites undergoing state-dependent changes in solvation, we were able to systematically confer temperature-sensing phenotype to a prototypical voltage-dependent potassium channel. We also demonstrate that magnitude of voltage-sensing charges inversely modulate temperature-sensitivity consistent with predictions of thermodynamic coupling. These emerging molecular principles provide a template to understand varied temperature-dependent gating phenotype in channels with conserved transmembrane architecture. 2145-Pos Board B282 The Gating Charge of Kv1.2 is Less than Expected from its Similarity to Shaker Itzel G. Ishida1,2, Gisela E. Rangel-Yescas2, Leon D. Islas2. 1 The Rockefeller University, New York, NY, USA, 2Facultad de Medicina, Departamento de Fisiologia, Universidad Nacional Autonoma de Mexico, Mexico City, Mexico. Much has been learned about the voltage sensors of ion channels since the Xray structure of the mammalian voltage-gated potassium channel Kv1.2 was published in 2005. The availability of high-resolution structural data of a Kv channel paved the way for the structural interpretation of numerous electrophysiological findings collected over many years in a variety of ion channels, most notably Shaker, and allowed the development of meticulous computational simulations of the activation mechanism of Kv1.2. The fundamental premise of the validity of the interpretation of functional measurements from Shaker using the structure of Kv1.2 is that both channels are related closely enough such that correlation of their data is a trivial task. We set out to confirm these assumptions by measuring the voltage sensitivity of the channel using the limiting slope method, followed by the determination of the gating charge through gating current recordings. We found that the gating charge, as measured by both techniques, is 10 e0, ~25% less than the wellestablished 13-14 e0 in Shaker. Next, we neutralized each of the six positive residues in S4 of Kv1.2 to probe the cause of the reduction of the gating charge, and found that while replacing R1 with glutamine decreased voltage sensitivity to just about 50% of the wild-type channel value, mutation of the subsequent arginines did not have an effect nearly as large. These data stand out as different to Shaker’s, where removal of the first four positive residues reduces the gating charge by roughly the same amount of elementary charges. We propose that the septum that separates the aqueous crevices in the VSD of Kv1.2 is thicker than Shaker’s, and that this accounts for a smaller gating charge in Kv1.2. Tuesday, February 10, 2015 2146-Pos Board B283 Discontinuity between the Voltage-Sensor and the Pore Domain does not Abolish Voltage-Gating of Kv10.1 Potassium Channel Adam P. Tomczak, Eva Lo¨rinczi, Juan Camilo Gomez-Posada, Walter Stu¨hmer, Luis A. Pardo. Molecular Biology of Neuronal Signals, Max Planck Institute of Experimental Medicine, Go¨ttingen, Germany. Voltage-gating of ion channels is crucial for excitable tissues, such as nerve and muscle. Here we show that a voltage-gated potassium channel retains its voltage-dependency of activation, even when the voltage sensor and the pore domain are expressed as two individual proteins from separate cRNAs in Xenopus laevis oocytes. Not only interrupting the S4-S5 cytoplasmic linker at various positions, but also concomitant deletion of several consecutive amino acids from this region yielded functional channels. Moreover, mutations of the voltage-sensor that shift the conductance-voltage curve in either hyperpolarizing or depolarizing direction cause the same shift when the S4-S5 linker is disrupted. Detailed characterization of how the location of discontinuity affects the voltage and time-dependence of activation and deactivation of the split constructs sheds new light on the coupling between the voltage-sensing module and the channel gate. Our findings indicate that an intact S4-S5 linker is not a sine qua non condition for voltage gating in Kv10.1. In consequence, the idea of direct mechanical coupling between the voltage sensor and the pore mediated by the S4-S5 linker needs to be revised, at least for the KNCH-family channels, which may have a different gating mechanism than Shaker. 2147-Pos Board B284 Two KCNQ1 Mutations Associated with Familial Atrial Fibrillation, S140G and V141M, Demonstrate Distinct Voltage Sensor Phenotypes Gary Peng1, Kevin J. Sampson1, Rene Barro-Soria2, H. Peter Larsson2, Robert S. Kass1. 1 Columbia University, New York, NY, USA, 2University of Miami, Miami, FL, USA. KCNQ1 is a voltage-dependent potassium channel that is expressed in the heart with the b-subunit KCNE1 to generate the slowly activating IKs current that plays a critical role in cardiac impulse conduction by allowing cardiac repolarization. Mutations in KCNQ1 leading to slowing of channel deactivation have been linked to arrhythmias including Short QT syndrome and familial atrial fibrillation. Two adjacent disease-linked mutations located in the voltage sensing domain (S1-S4) of KCNQ1, S140G and V141M, have been shown to drastically slow current deactivation. While their effects on IKs current deactivation kinetics are similar, their mechanisms may differ. For example, in the absence of KCNE1, S140G but not V141M slows current deactivation. Moreover, crosslinking studies suggest that while V141M can directly interact with KCNE1, S140G cannot. We explore the hypothesis that S140G and V141M, while exhibiting similar effects in IKs current gating, demonstrate distinct phenotypes on voltage sensor movement. Using voltage clamp fluorometry, we studied voltage sensor movement simultaneously with channel current. We found that in the absence of KCNE1, S140G but not V141M slows voltage sensor deactivation, consistent with their effects on current. Furthermore, in the presence of KCNE1, S140G slows voltage sensor movement, but V141M does not slow voltage sensor deactivation. This work shows that while S140G slows both the current deactivation and voltage sensor movement in the presence and absence of KCNE1, in contrast, V141M slows current deactivation only in the presence of KCNE1, without significantly slowing the kinetics of voltage sensor movement. This suggests that these two mutations slow the deactivation of KCNQ1/KCNE1 channels by different mechanisms, an observation made possible by the simultaneous measurement of both voltage sensor movement and channel current. 2148-Pos Board B285 Molecular Determinants of Voltage Sensor Domain Activation Lucie Delemotte1, Vincenzo Carnevale1, Michael L Klein1, Marina A. Kasimova2, Mounir Tarek2. 1 Chemistry, Temple University, Philadelphia, PA, USA, 2Chemistry, Universite de Lorraine, Nancy, France. The voltage sensor domain (VSD) is a four transmembrane segments protein domain that confers voltage sensitivity to ion channels and other proteins. The VSD senses changes in the external potential through its highly positively charged fourth transmembrane segment (S4). The activation mechanism involves a complex, helical screw motion of S4 during which the salt bridge pattern between the S4 arginines and the negative charges of S1-S3 and of the lipid headgroups reorganizes. Together, this ratchet-like motion brings the VSD from the resting to the activated state in a series of jumps that proceed through several intermediate states. We use molecular dynamics simulations 427a and free energy calculations to characterize the free energy landscape and the kinetic rates associated with the different steps of the activation process. To highlight the molecular determinants of this complex conformational transition, we apply techniques of machine learning and data analysis. Specifically, a custom-tailored dimensionality reduction approach is used to extract the relevant degrees of freedom describing the concerted motion of protein residues, lipids and waters. 2149-Pos Board B286 Role of the Voltage Sensing Domain S1-S4 in TRPV1 Channels Juan Zhao1, Rikard Blunck2. 1 Departments of Physics and Physiology,, Groupe d’E´tude des Prote´ines Membranaires (GE´PROM), Universite´ de Montre´al, Montreal, QC, Canada, 2 Department of Physics and Physiology,, Groupe d’E´tude des Prote´ines Membranaires (GE´PROM), Universite´ de Montre´al, Montre´al, QC, Canada. Transient Receptor Potential Vallinoid type 1 (TRPV1) channel is a voltage-, heat-, and ligand-activated, nonselective cation channel. As a member of hexahelical cation channel superfamily, TRPV1 does not feature the positively charged S4, which is responsible for the voltage sensitivity of voltage-gated ion channels (VGICs). The origin for TRPV1’s voltage sensitivity must lie elsewhere. The objective of this study was to investigate the role of ‘‘classical’’ S1-S4 voltage sensor, in particular S4, in the TRPV1, and obtain molecular information about the functional coupling between voltage, chemical and temperature sensors. To test whether the positive charge is involved in activation and to explore the function of the S4 segment in TRPV1 activation, we made a series of single point arginine replacements that scanned residues from E536 through T556 in the S4 and characterized their response to capsaicin, protons and heat using cut-open oocyte voltage-clamp. We also constructed TRPV1/ Shaker chimeras in which the S1-S4, S3-S4, or S4 segments of the TRPV1 channel were replaced by the corresponding segments of Shaker channels. The introduction of positively charged residues in the S4 segment resulted in a steeper activation curve and a defective response to capsaicin and protons. Importantly, several of the mutant channels displayed strong inhibition of hyperpolarization-activated inward currents. Three chimeras gave rise to functional channels that exhibited stronger voltage dependence at positive voltages than TRPV1. The chimeras progressively lost inward current with increasing portions replaced by the corresponding Shaker region. They were activated by protons with higher sensitivity compared with TRPV1. These results suggest that re-established outward rectification was dominant over other gating stimuli; in other words, keeping the S1-S4 in the ‘‘deactivated’’ state prevented activation by protons, capsaicin or temperature. In wildtype TRPV1, the S4 seems to inhibit activation by protons. 2150-Pos Board B287 Sensing the Electrochemical KD Gradient: The Voltage Gating Mechanism in K2P Potassium Channels Marcus Schewe1, Ehsan Nematian-Ardestani1, Thomas Linke2, Klaus Benndorf2, Stephen J. Tucker3, Markus Rapedius1, Thomas Baukrowitz1. 1 Department of Medicine, Institute of Physiology, Kiel, Germany, 2 Department of Medicine, Institute of Physiology II, Jena, Germany, 3 Department of Physics, Biological Physics Group, Clarendon Laboratory, Oxford, United Kingdom. Two-pore domain (K2P) Kþ channels represent a large family of ion channels that are major regulators of cellular excitability in the body and involved in a wide range of cellular mechanisms including apoptosis, vasodilatation, anaesthesia, pain, neuroprotection and temperature sensing. Many K2P channels are strongly activated by membrane depolarization, but the mechanisms underlying this voltage-dependent behaviour are unknown. Here we report that many K2P channels (e.g. TREK-1, TREK-2, TRAAK, TASK-3, TALK-2 and TRESK) are equipped with a gating machinery which directly senses the electrochemical Kþ gradient and which gates the pore open when the membrane potential is positive to the Kþ reversal potential. These properties couple voltage activation in K2P channels tightly to the reversal potential in distinction to classical Kv channels. We show that this sensing mechanism is located in the selectivity filter (SF), is strongly affected by the permeant ion species and operates as a check valve that is opened by outward permeation but closed by inward permeation. This gating behaviour is steeply voltage-dependent suggesting that multiple ions within the SF are moved simultaneously by the electrical field to gate the filter open. These findings highlight a mechanism of voltage-dependent gating which bypasses the need for electromechanical coupling to a separate voltage sensing module and is instead powered directly by the electrochemical gradient. This also further closes the mechanistic gap between ion channels and transporters because in both cases the electrochemical gradient is used to power a conformational change leading to either an ion 428a Tuesday, February 10, 2015 translocation step in the case of a transporter or a pore gating step in K2P channels. 2151-Pos Board B288 Can ClC-2 Chloride Channel be Activated by Hyperpolarization Alone in Cells Dialyzed with Non-Permeant Anions? Jose´ J. De Jesu´s-Pe´rez1, Alejandra Castro-Chong1, Ru-Chi Shieh2, Carmen Y. Herna´ndez-Carballo1, Jose A. De Santiago-Castillo3, Jorge Arreola1. 1 Institute of Physics, Univ. Autonoma de San Luis Potosi, San Luis Potosi, Mexico, 2Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan, 3 ReliaXpert, San Luis Potosi, Mexico. Open and closing of ion channels is driven by conformational changes triggered by either an intrinsic voltage sensor in voltage-gated channels or by ligand binding in ligand-gated channels. However, ClC-2, a two pore Cl channel is activated by hyperpolarisations despite of lacking of an intrinsic voltage sensor. The structural information available to date strongly suggests that in the closed conformation each pore of ClC-2 is occluded by the negatively charged side chain of a glutamate residue. Although the gating mechanism is unknown, there is evidence which indicates that intracellular anions and extracellular protons regulate gating. The present work was aimed to determine the contribution of pore occupancy caused by intracellular anions in gating the mouse ClC-2 channel. In our experiments, ClC-2 was readily activated by hyperpolarisations when permeant anions such Cl, SCN, Br and I were present on either side of the membrane. However, ClC-2 was not activated in cells dialyzed with acetate (0 Cl) and exposed to [Hþ]O¼107.3 M. In contrast, the channels were opened by increasing [Cl]i at [Hþ]O¼1010 M_a condition unlikely to protonate the glutamate’s side chain. Importantly, voltage gating occurred when F or glutamate were present in the cytosolic side. Since these two anions are non-permeant we propose that they must enter the pore and interact with the glutamate side chain from the inside in order to induce opening. Thus, we propose that a strong hyperpolarisation drives the intracellular anions into the permeation pathway and the subsequent electrostatic interaction between the anions and the negatively charged side chain opens the pore. Our data support the hypothesis that voltage-dependent gating in mouse ClC-2 requires intracellular anions. Supported by grant 219949 from CONACyT. 2152-Pos Board B289 Molecular Basis of Voltage-Dependent Gating in ClC Transporters Jan-Philipp Machtens1, Matthias Grieschat2, Christoph Fahlke1, Alexi K. Alekov2. 1 Institute of Complex Systems, Zellula¨re Biophysik (ICS-4), Forschungszentrum Ju¨lich, Ju¨lich, Germany, 2Institut fu¨r Neurophysiologie, Medizinische Hochschule Hannover, Hannover, Germany. The CLC family encompasses Cl channels and coupled Cl/Hþ exchangers. CLC channels and transporters both exhibit voltage-dependent gating, the physiological importance of which is illustrated by multiple diseasecausing mutations that specifically result in altered voltage sensing. Despite a large body of available functional and structural data, the molecular mechanisms underlying voltage gating in CLCs still remain unclear. Using electrophysiological admittance measurements, we recently decomposed voltage-dependent activation of the human ClC-5 transporter into multiple discrete electrogenic transitions (Grieschat M & Alekov AK (2014) Biophys J 107, L13–L15). A key player of the gating machinery appears to be the socalled gating glutamate (Gluext; Glu148 in EcClC), which is thought to move upon changes in voltage. To better understand the basis of voltage-dependent gating, we conducted molecular dynamics simulations of the bacterial homologue EcClC. Using a double-bilayer system, we applied small ionic concentration gradients across the membrane that resulted in charge imbalances Dqion and we calculated the resulting potential differences Vm. Analysis of the dependence of Vm on Dqion for various conformations that differed in Cl binding site occupancy and the protonation state of Gluext was used to determine charge displacements of the transporter along the electric field. Multiple processes, including protonation and conformational changes of Gluext, binding of Cl ions and refocusing of the electric field triggered by changes in water accessibility are associated with charge transfer across the membrane and therefore exhibit intrinsic voltage dependence. We calculated gating charges that underlie these conformational transitions of the CLC transporter. Based on these calculations, gating charge recordings and capacitance measurements on ClC-5, we propose a molecular description of CLC voltage sensing which attributes fractional gating charges to these partial reactions of the transport cycle. 2153-Pos Board B290 Multiphasic Profiles: Discontinuous Transitions in Conductance-Voltage Data for Ion Channels Per Nissen. ˚ s, Norway. Norwegian Univ. of Life Sciences, A Multiphasic profiles, a series of straight lines separated by discontinuous transitions, were first found (Nissen, Annu. Rev. Plant Physiol. 1974) for the concentration-dependence, plotted in linear transformations of the MichaelisMenten equation, for ion uptake in plants. Reanalyses of recently published data show that conductance-voltage data for ion channels are also well represented as multiphasic. In contrast, the Boltzmann function and other functions giving curvilinear profiles must often be rejected for statistical reasons (very low probabilities by the Runs test that the uneven distribution of points around the profiles is due to chance). Plots of deviates show that the fits to multiphasic profiles are usually better than the fits to curvilinear profiles, i.e. that the data are more precise than apparent from the published plots. As shown by simulation, the better fits to multiphasic profiles are not due to errors (noise) in the data. Adjacent lines are often parallel. The transitions are then necessarily in the form of noncontiguities (jumps). Non-parallel lines are also frequently separated by jumps. Most often, replicate determinations give different multiphasic patterns (number of phases, voltages at which the transitions occur). The use of the averages will then give a meaningless pattern. Whenever the data are sufficiently detailed and precise, multiphasic profiles are also found for a variety of other processes and systems, biological as well as nonbiological: 1) pH profiles, including profiles for non-enzymatic model systems. 2) Binding as determined by fluorescence anisotropy titration or isothermal titration calorimetry (ITC). 3) Folding and unfolding of proteins. Implications for the opening and closing of single biological channels will be discussed. 2154-Pos Board B291 A Highly Cooperative and Steeply Voltage Gated Channel Triplet Shang H. Lin1, Benjamin Wu2, Marco Colombini2. 1 University of Maryland, College Park, Greenbelt, MD, USA, 2University of Maryland, College Park, College Park, MD, USA. When reconstituted into planar phospholipid membranes, a 4.5nS (in 1M KCl) membrane channel complex behaves as if it is composed of three channels that are operating in a highly cooperative manner. The channels are steeply voltagegated (e-fold for 1.8 mV) and well-organized. In the 5 70mV range, no voltage gating takes place until the first channel, channel 1, closes at high positive potentials (>70 mV). Only following this closure does channel 2 begin to gate in response changes in voltage. For channel 2, closure takes place at low negative potentials (typically 25 to 30 mV). It is only when channel 2 is closed that channel 3 closes routinely but does so at low positive potentials (typically 25-30 mV). Simultaneous reopening of channels 2 and 3 is common, demonstrating especially high cooperativity. These and other remarkable properties support a working model for the gating and cooperativity of these channels. The highlights include anti-parallel orientation of the channels and the voltage sensor dipole being responsible both for voltage gating and cooperativity. (Supported by NSF grant MCB-1023008) Tuesday, February 10, 2015 Ligand-gated Channels II 2155-Pos Board B292 Non-Equivalent Ligand Selectivity of Agonist Sites in (a4b2)2a4 Nicotinic Acetylcholine Receptors: A Key Determinant of Agonist Efficacy Simone Mazzaferro, Federica Gasparri, karina New, Constanza Alcaino, Isabel Bermudez. Biological and Medical Sciences, Oxford Brookes University, Oxford, United Kingdom. The a4b2 nicotinic acetylcholine receptor (nAChR) is the most abundant nAChR type in the brain, and this type plays a pivotal role in the reinforcing and rewarding effects of nicotine. The a4b2 nAChR exists in alternate (a4b2)2 a4 and (a4b2)2b2 forms, which are activated by agonists with strikingly differing efficacies. Recent breakthroughs have identified an additional operational agonist binding site in the (a4b2)2a4 nAChR that increases the maximal responses to ACh. In this study, we characterised the ligand selectivity of the individual agonist sites of the (a4b2)2a4 nAChR to determine whether differences in agonist selectivity influences agonist efficacy. Applying the substituted cysteine accessibility method to individual agonist sites in concatenated (a4b2)2a4 receptors, we determined the agonist selectivity of the agonist sites of the (a4b2)2a4 receptor. We show that (a) the rate of covalent modification of substituted cysteines depends on the subunit interface at which the modification occurs and b) that agonists such as sazetidine-A and TC-2559 are excluded from the site at the a4/a4 interface. Given that additional binding to the agonist site in the a4/a4 interface, increases ACh efficacy and that agonists excluded from the agonist site at the a4/a4 interface behave as partial agonists, we conclude that the ability to engage all agonist sites in (a4b2)2a4 nAChRs is a key determinant of agonist efficacy. The findings add another level of complexity to the structural mechanisms that govern agonist efficacy in heteromeric nAChRs and related ligand gated ion channels. 2156-Pos Board B293 Desformylflustrabromine Potentiates High-Sensitivity a4b2 Receptors by Increasing Channel Opening Rate Arianna Demmerly, Brian W. Edmonds. Chemistry & Biochemistry, University of Alaska Fairbanks, Fairbanks, AK, USA. Desformylflustrabromine (dFBr) is a positive allosteric modulator of a4b2 neuronal nicotinic receptors (nAChRs). Investigations of mechanisms of nAChR modulators may facilitate the development of new therapeutic drugs that offset or correct errors in signaling associated with Alzheimer’s disease, nicotine addiction, and other disorders of cholinergic signaling. We made steady-state, cell-attached patch-clamp recordings of 20 pS, high-sensitivity (HS) human a4b2 receptors stably expressed in HEK-293 cells. We developed 6-state gating models from data acquired with 1 mM ACh with and without 1 mM dFBr. Models included two sequential ligand-binding reactions leading to a desensitized state (C14C24C34D), and mono- and diliganded openings (O1 and O2) from C2 and C3, respectively. Rate constants governing transitions between connected states were obtained by fitting single-channel data to the model (constraints: C1/C2 ¼ 2,C2/C3 and C3/C2¼2,C2/C1) using a maximum likelihood method implemented in QUB. dFBr increased binding and unbinding rates for ACh (2.6- and 2-fold, respectively), the opening rate (C3/O2) for diliganded receptors (4.8-fold), and the (microscopic) desensitization rate (5.3-fold). Simulations of macroscopic responses to concentration-jumps (range: 0.1 mM - 100 mM) revealed that the enhanced rate into O2 yielded approximately 2 to 3-fold potentiation of peak currents (maximal Popen in 100 mM ACh ¼ 0.29) despite the fact that potentiation was blunted by the enhanced rate of desensitization. Moreover, at ACh concentrations above 10 mM, our data are consistent with the interpretation that potentiation of HS receptor responses to dFBr arises not from modulation of ligand binding rates, but from an increase in the rate of channel opening, corresponding to a decrease in the latency to first opening and a reduction in the time to peak current (ACh 23 ms; dFBr 8 ms) of simulated macroscopic responses. 2157-Pos Board B294 Is the AChR a Cuckoo-Clock? Local and Remote Interactions of the a M23 Linker in Gating Shaweta Gupta, Prasad Purohit, Anthony Auerbach. Physiology and Biophysics, University at Buffalo, Buffalo, NY, USA. The aM2-3 linker of pentameric ligand-gated ion channels (18-residues in AChRs) is at the interface of the extracellular and transmembrane domains, which twist relative to each other early in the channel-opening process (phi~0.7). Phi-analyses of mouse muscle AChRs indicate that residues in this linker and those at the transmitter binding sites share a common, even-earlier 429a position in the gating transition state (phi~0.9). To understand the mechanism ˚ ), whose combined for the similar behavior of these two separated regions (~30 A motions appear to trigger channel-opening, we used single-channel free-energy mutant cycle analyses (A-A pairs) to measure the extents to which a-subunit linker residues (265-268) interact with other amino acids. The significant interactions (>0.5 kcal/mol) were as follows (n pairs tested; energies in kcal/mol): i) aloop2 (7), aE45-aP265 ~þ3.5 (positive is unfavorable); ii) acys-loop (2), aV132-aS268 ~þ1; iii) agonist sites (3), aS268 and aY93 or aY198 showed no coupling but the interaction with aY190 was ~-1.2 (favorable); iv) intra-linker (4), no significant coupling; v) εloop9 (6), εG183-aS268 and εG183-aT267 ~þ1.4; vi) εpre-M1 (1), εL221-aT267, no coupling. The large, un˚ favorable interaction between aP265 and its loop 2 neighbor aE45 (~5 A separation in GLIC) suggests that these side chains share a common energetic environment that is perturbed by the gating conformational change (twist). The long-distance, favorable interaction between aS268A in the linker and aY190A at the agonist site is more surprising. We hope to elucidate a network of apposed residues whose shared energy changes might be associated with the twist. More importantly, we are exploring the possibility that there is a longdistance energy transfer between the agonist binding site and transmembrane domain via the backbone that is not by a cuckoo-clock-like mechanical linkage. 2158-Pos Board B295 Molecular Simulations of Muscle AChR Agonist Binding Sites Srirupa Chakraborty1, Tapan K. Nayak1, Iva Bruhova1, Wenjun Zheng2, Anthony L. Auerbach1. 1 Biophysics, State University of New York at Buffalo, Buffalo, NY, USA, 2 Physics, State University of New York at Buffalo, Buffalo, NY, USA. We are attempting to design and control the agonist binding properties of muscle acetylcholine receptors (AChRs). These hetero-pentamers have 2 transmitter binding sites in the extracellular domain, at a-d and either a-g (fetal) or a-ε (adult) subunit interfaces. We used computational modeling and MD simulation to investigate the structural dynamics and energetics of these 3 different binding interfaces. Starting from an Aplysia AChBP crystal structure (PDB id 2BYQ), we used homology modeling to build all-atom dimer and pentamer models of the extracellular domain, and docked acetylcholine (ACh) as the ligand. MD simulations (Charmm27) were run to obtain binding free energies at all 3 interfaces. The a-d and a-ε sites were found to be approximately equivalent, but a-g provided ~33% more favorable binding energy, which is in good agreement with experimental, single-channel electrophysiology results (Nayak and Auerbach, PNAS 2013). In the model, the packing of five aromatic residues is tighter, the tryptophan residues 149 and 55 are more orthogonal and loop C less is flexible at a-g compared to the others. The dimer and pentamer models gave similar binding energies, supporting the experimental observation that the agonist sites behave independently. We are using the model as a tool, to identify residues in the ε and d subunits that are responsible for the lower ACh binding energy and, hence, to guide the wet-bench experiments. We targeted nine amino acids that are homologous in d and ε but different in g, within 20 ˚ of the quaternary ammonium of ACh. In the pentamer simulations, swapping A all of these between a-d and a-g resulted in a full switch of the structures and energies. We are testing various subsets of these mutations in order to suggest a minimum construct that can be verified by using electrophysiology. 2159-Pos Board B296 Differences in Agonist Energy at the Neurotransmitter Binding Sites in the Neuromuscular Acetylcholine Receptors Tapan K. Nayak, Anthony Auerbach. Physiology and Biophysics, SUNY @ Buffalo, Buffalo, NY, USA. Each neuromuscular AChR has 2 agonist binding sites located in the extracellular domain, at ad and either aε (adult) or ag (fetal) subunit interfaces. Though in most species g- is replaced by the ε-subunit during perinatal development, the rationale for the subunit swap is little understood. We used singlechannel electrophysiology to measure the effects of mutations of 5 conserved aromatic residues at each type of agonist site, with regard to their contribution to the free energy of agonist binding to active vs. resting receptors (DGB1; in mouse AChRs with only 1 functional binding site expressed in HEK cells). The agonist binding sites in both adult and fetal AChRs operate independently. For 4 different agonists (including ACh and choline) DGB1 is ~-2 kcal/mol more favorable at ag compared to aε and ad. Of the aromatic residues, only 3 contribute to the DGB1 at the adult sites (aY190, aY198 and aW149), whereas all 5 do so at ag (including aY93 and gW55). The non-a tryptophan (W55) is a variable energy source that makes a huge contribution to DGB1 only at ag, where it shapes the binding pocket by interacting with some of the a-subunit aromatics. The hydroxyl and benzene groups of loop C residues aY190 and aY198 behave similarly at all three kinds of site, but the benzene of aY93 contributes to DGB1 only at ag. We are studying other residues on the non-a side of 430a Tuesday, February 10, 2015 the pocket for their contribution to DGB1 to understand the interactome network at the three binding sites. It is possible that the different sensitivities of the fetal a-g site vs. the adult a-ε and a-d sites to ACh and choline are important for the proper maturation and function of the neuromuscular synapse. 2160-Pos Board B297 Interaction of 7-Methoxytacrine-Adamantylamine Cholinesterase Inhibitors with Nicotinic and Muscarinic Acetylcholine Receptors Ze-Jun Wang1, Vendula Sepsova2, Katarina Spilovska2, Tasnim S. Mohamed1, Ayman K. Hamouda1,3. 1 Dept. of Pharmaceutical Sciences, Texas A&M Health Science Center, Kingsville, TX, USA, 2Dept. of Toxicology, University of Defense, Hradec Kralove, Czech Republic, 3Dept. of Neuroscience and Experimental Therapeutics, Texas A&M Health Science Center, Bryan, TX, USA. Acetylcholinesterase inhibitors (AChEI) and the N-methyl-D-aspartate (NMDA) receptor antagonist memantine are among the few FDA-approved drugs for Alzheimer’s disease. In effort to develop multi-target (AChENMDA)-directed ligands for the treatment of Alzheimer’s disease, a series 7Methoxytacrine-Adamantylamine thiourea heterodimers have been synthesized and evaluated for their effects as AChEI (Spilovska et. al. 2013, Molecules 18, 2397-2418). In this report, we extend their pharmacological characterization by examining their effects on neuronal and muscle-type nicotinic acetylcholine receptors (nAChR) and muscarinic acetylcholine receptors (mAChR). We also compare their pharmacology to 7-Methoxytacrine-Adamantylamine urea heterodimers. 7-Methoxytacrine-Adamantylamine thiourea derivatives containing 2-8 carbon linkers inhibited AChE and butyrylcholinesterase (BChE) with high potency (IC50, 0.5-6 mM), whereas 7-Methoxytacrine-Adamantylamine urea derivatives inhibited AChE with higher potency than BChE. None of the tested compound potentiated M1 mAChR or nAChRs responses. They have minimal effects on acetylcholine-induced currents in Xenopus oocytes expressing neuronal a4b2 nAChR and inhibited M1 mAChR at higher concentrations (IC50s >10 mM). At concentration that inhibit AChE, both thiourea and urea derivatives inhibited acetylcholine-induced currents in Xenopus oocytes expressing muscle-type AChRs. Our preliminary results also suggest that the length of the carbon linker affect 7-Methoxytacrine-Adamantylamine urea interactions with AChRs, and further modifications of their structure are expected to yield compounds with more favorable pharmacological profiles. 2161-Pos Board B298 Stepchild Nicotine: Effect of the Name-Giving Agonist on Muscle-Type Nicotinic Acetylcholine Receptor Abhilasha Ladha, Klaus Benndorf, Jana Kusch. Institute of Physiology II, University Hospital, Jena, Germany. Muscle-type nicotinic acetylcholine receptors (nAChRs) mediate fast synaptic cholinergic responses at neuromuscular junctions. The adult subtype of the receptor is composed of a1, b1, d, and ε subunits in the ratio 2:1:1:1. Although nicotine is the name-giving agonist for ionotropic acetylcholine receptors, only a few studies investigated its effect of nicotine on this specific nAChR subtype. A comprehensive knowledge of the mechanism underlying the nicotineinduced gating is still missing. For this reason, we had a closer look to macroscopic (a1)2b1dε receptor currents evoked by fast nicotine jumps and compared them with acetylcholineevoked responses. The receptors were heterologously expressed in HEK293 cells. After lifting and positioning the cells in front of a piezo-driven doublebarreled application pipette, whole-cell currents were monitored. Herein, nicotine was found to be an agonist with a relative potency of about 0.60 compared to the full agonist acetylcholine. The efficiency was about 30 times lower than for acetylcholine (EC50 ¼ 97.0 mM versus EC50 ¼ 3.35 mM). When activated by nicotine, (a1)2b1dε receptors desensitize slower than receptors activated by acetylcholine. As expected from single-channel analysis (Akk and Auerbach, Br J Pharmacol. 128:1467-76, 1999; Jadey et al., J Gen Physiol. 141:95-104, 2013), off-currents, occurring after removal of high nicotine concentrations, confirmed the idea of nicotine acting as a channel blocker at concentrations higher than 1 mM. In summary, the data provide new insight into the nicotine-induced gating of adult muscle-type receptors. The differences in comparison to the physiological ligand acetylcholine and the action of nicotine as a partial agonist are discussed. 2162-Pos Board B299 The Role of the M3 Helix in AChR Gating and PAM Action Aashutosh Vihani, Prasad Purohit, Anthony Auerbach. University at Buffalo, Buffalo, NY, USA. We are examining the M3 transmembrane helix of mouse muscle AChRs with regard to C4O gating and the effects of positive allosteric modulators (PAMs). The relative ground and transition state gating energies were estimated from single-channel currents of AChRs having M3 mutations. Diliganded gating rate and equilibrium constants were measured for ~100 mutations in 5 subunits (a, b, d, ε and g; 20 mM choline or 500 mM ACh, 100 mV, HEK cells). In general: i) the effects of the mutations were small (%1 kcal/mol), ii) the effects were similar among subunits, iii) the mutations in different subunits were independent, iv) the mutations have no effect on the energy from the agonist affinity change (%~0.3 kcal/mol) and v) most phi values were low (~0.3). Crystal structures of AChR homologues in detergents suggest that the low phi positions in M3 are interaction sites for PAMs. We investigated the actions of PNU-120596 and IVM (1-1000nM). In WT AChRs (adult and fetal) these ligands increase substantially the frequency and lifetime of constitutive openings in a manner consistent with action at a low-phi position such as in M3. Such potentiation was also apparent at low [agonist] (<1mM ACh or choline), but at higher [agonist] gating kinetics became complex and the potentiation was less clear. The PAMs also increased substantially the frequency and lifetime of constitutive openings in some of the M3 mutants (to W). 2163-Pos Board B300 Modal Gating of Muscle AChRs having Loop C Mutations Ridhima Vij. SUNY Buffalo, Buffalo, NY, USA. Many ion channels exhibit multiple patterns of kinetic activity (‘modes’) in single-channel currents. This behavior is rare in WT mouse muscle acetylcholine receptors (AChRs), where A2C4A2O gating events are well-described by single exponentials. We have found that mutations of all loop C residues at the transmitter binding site (except aY190 and aY198) increase the probability of modes. The free energy of diliganded gating is the sum of that for unliganded gating and from agonist affinity-changes at 2 binding sites: DG2¼DG0þ2DGB. For WT adult AChRs these values are (100 mV; kcal/mol): 1.9, þ8.3 and 5.1 (ACh). All mutations of aP197 (ACDEGIKSVY) had at least 2 gating modes with all agonists (ACh, Cho, CCh and TMA). We focused on aP197A and ACh and Cho. There were 3 distinct modes at saturating [agonist] but only 1 mode without agonist. Hence, the kinetic heterogeneity for this construct is generated by fluctuations in DGB. DDGB was estimated for each mode (kcal/mol) for ACh (1.7, WT and þ1.6) and Cho (2.2, WT and þ0.9). That is, the gating equilibrium constant for one mode was ~30-times larger than the WT (for both agonists), for another was about the same as the WT and for the third was ~8-times smaller. An added F mutation at aY190 or aW149 reduced modal gating whereas one at aY198 or aY93 did not. An F substitution at εW55 had no effect, but one at dW57 reduced the modes. We also examined AChRs having only 1 active binding site and observed less heterogeneity from aP197A at the fetal a-g site vs. the adult a-ε or a-d sites. Our working hypothesis is that mutations in loop C allow the agonist to sample multiple, meta-stable positions within the binding pocket, to produce distinct DGB values. 2164-Pos Board B301 Dissecting the Inhibitory and Potentiation Effects of Desformylflustrabromine on a Muscle-Type Nicotinic Acetylcholine Receptor (NACHR) Tasnim S. Mohamed1, Ze-Jun Wang1, Tiffany R. Trevino1, Ayman K. Hamouda1,2. 1 Pharmaceutical Sciences, Texas A&M Health Science Center, Kingsville, TX, USA, 2Dept. of Neuroscience and Experimental Therapeutics, Texas A&M Health Science Center, Bryan, TX, USA. Desformylflustrabromine (dFBr) potentiates 4b2 nAChR in vitro (Kim et al. 2007, Biorg. Med. Chem. Lett. 17: 4855) and reduces nicotine selfadministration in vivo (Liu 2013; Psychopharmacology 230: 203). We have reported that dFBr is a potent inhibitor (IC50, ~1 M) of human (Ha12b1εd) and Torpedo (Ta12b1gd) muscle-type nAChRs. We also found, using [3H]dFBr reversible binding and dFBr displacement of [3H]PCP binding, that dFBr binds with high affinity to the muscle-type nAChRs ion channel in the desensitized state. But, when we photolabeled nAChR-rich Torpedo membranes with [3H] dFBr in the presence of agonist, four dFBr binding sites were evident. [3H] dFBr photolabeled amino acids in the ion channel and in three binding sites within the extracellular domain previously identified for nAChRs PAMs galanthamine and physostigmine. The high affinity binding of dFBr in the ion channel precludes the ability to study the functional contribution of dFBr binding within the extracellular domain and to determine whether dFBr acts as inhibitor or potentiator at these sites. Therefore, we are introducing mutations in the muscle-type nAChR ion channel M2 helices to eliminate the channel blocking effect of dFBr. The effect of M2 mutations on the EC50 of ACh and on dFBr inhibition of muscle-type nAChR were examined using two-electrode voltage clamp recording. Serine substitutions of the leucine at M2-9 of each nAChR subunit decreased dFBr IC50 compared with wild-type, consistent with an open channel blocking mechanism. This results confirm our [3H]dFBr the photolabeling results demonstrating that Tuesday, February 10, 2015 dFBr binds within the ion channel in close proximity to M2-9. The effects of other substitutions at M2-9 and at other positions in the M2 helices on dFBr inhibition of muscle-type nAChR are being tested. 2165-Pos Board B302 Exploring Alpha7 Positive Allosteric Modulators from a Single-Channel Perspective Natalia D. Andersen1, Jeremias Corradi1, Fernanda Tolosa1, Nehuen Gasparini1, Hugo R. Arias2, Cecilia B. Bouzat1. 1 UNS/CONICET, Instituto de Investigaciones Bioquimicas de Bahia Blanca, Bahia Blanca, Argentina, 2Department of Medical Education, California Northstate University College of Medicine, Elk Grove, CA, USA. Through the enhancement of endogenous ACh responses, positive allosteric modulators (PAMs) of the nicotinic a7 receptor represent a promising therapeutic approach for the treatment of several neurological disorders. We combine single-channel and macroscopic current recordings to explore the molecular basis underlying potentiation of human a7 by three amide derivatives compounds named as Compound 2 (3-furan-2-yl-N-p-tolyl-acrylamide), 3 and 4 (5-100 mM). In contrast to the brief and isolated openings typical of a7, in the presence of Compounds 2, 3 or 4 opening events show significantly increased durations (~7-fold) and appear grouped in bursts of about 10-20 ms. This activity pattern is similar to that observed in the presence of 5-hydroxyindole (5-HI), a type I PAM, which leads to openings of ~2 ms and bursts of ~5 ms. PNU-120596, a type II PAM, produces a more profound increase in open duration, and opening events appear in long clusters that last for several seconds. 5-HT3A and a7-5HT3A receptors as well as an a7 quintuple mutant receptor that is insensitive to PNU are not potentiated by Compound 2. At the macroscopic level, Compound 2 and 5-HI increase the net charge (~5-fold and 8-fold at 50 mM and 2 mM, respectively). However, such increase is mainly due to the decrease in the decay rate for Compound 2, which resembles the effect of a type II PAM, and to the increase of the maximal peak currents for 5-HI. Our results demonstrate that these novel compounds may bind to the same intrasubunit transmembrane cavity as PNU, decrease desensitization as type II PAMs, and modify single-channel activity similarly as type I PAMs. They also contribute to elucidating the foundations of a7 modulation. 2166-Pos Board B303 Gating Ritual: Simulations of Gating in Glutamate-Gated Chloride Channel Ozge Yoluk1,2, Stephanie Heusser3,2, Magnus Andersson1,2, Laura Orellana1,2, Erik Lindahl1,2. 1 KTH, Stockholm, Sweden, 2SciLifeLab, Solna, Sweden, 3Stockholm University, Stockholm, Sweden. Ligand-gated ion channels (LGICs) are responsible for converting chemical signals to electrical ones in the nerve system, which is a key element in the fast synaptic signal transmission. The first crystal structures of prokaryotic homologs were solved a few years ago, and today there are many more structures available - including a human one. These structures are believed to represent both open, closed, and desensitized states, but despite this wealth of information the core question for a LGIC remains unanswered: How does the binding of the agonist couple to the transmembrane domain to cause gating? Recent crystal structures of GluCl in both apo and liganded states make GluCl an ideal model system to explore this coupling computationally. We have performed several microsecond-scale simulations of open and closed state crystal structures (3RHW, 3RIF, 4TNV ) in their native forms, and examined the effects of keeping & removing ligands as well as allosteric modulators to observe transitions events. Removal of both ligand and modulator leads to a fully closed state after more than a microsecond. Control simulations of open states (3RHW & 3RIF) display a closed hydrophobic gate (9’) at least half of the time. In 3RIF simulation, the 9’ position is more flexible and an open pore is reached within a microsecond. Although rest of the pore is open, in 3RHW such opening at 9’ almost never happens. In the open 3RIF simulations, the pore opening is correlated with an increased distance between subunits. Our simulations provide clear evidence of the coupling in dynamics between the extracellular and transmembrane domains of LGICs on microsecond scale, and show how the extracellular ligand binding affects both local structure and the pore gating more ˚ away in the transmembrane domain. than 50A 2167-Pos Board B304 Aromatic Residues in the Transmembrane Helices Play an Essential Role in the Homopentameric Assembly of the Glucl a Anke Dopychai, Clairentine F. Pokam, Gunther Schmalzing. Molecular Pharmacology, RWTH Aachen University, Aachen, Germany. Pentameric ligand-gated ion channels (pLGICs) represent a major class of fast neurotransmitter receptors. In addition to nicotinic acetylcholine receptors 431a (nAChRs), glycine receptors, GABAA receptors and 5HT3 receptors of vertebrates, pLGIC homologs have been identified in invertebrates and even in bacteria. Several crystal structures of pLGIC homologs have been solved at high resolution. By combining alanine scanning mutagenesis and homology modeling based on the X-ray structure of ELIC and the electron microscopy structure of the muscle-type nAChR, we could previously show that a network of aromatic residues within the transmembrane domains TM1, TM3 and TM4 is important for the proper assembly of human glycine receptor protomers into a functional homopentamer (Haeger et al. (2010), Nat Struct Mol Biol 17: 90-98). Here, we examined whether the TM helices of pLGIC homologs with solved crystal structure are also involved in homopentameric assembly. We chose the glutamate-gated chloride channel (GluCl) a, a pLGIC from the nematode Caenorhabditis elegans, which is most similar to the a1 glycine ˚ resolution (Hibbs and receptor and whose crystal structure was solved at 3.3 A Gouaux (2011), Nature 474: 54-60). We performed an alanine scanning mutagenesis of all four TMs of GluCl a, purified the expressed GluCl mutants from Xenopus laevis oocytes by non-denaturing Ni2þ-NTA chromatography, and analyzed them by blue native PAGE and SDS-PAGE. We found that aromatic residues in TM1, TM3 and TM4 are important for the correct homopentameric assembly of GluCl a protomers. These findings indicate that the role of aromatic residues in the assembly of chloride-conducting pLGICs is conserved across species. 2168-Pos Board B305 Opening and Selectivity of the Glic Ligand-Gated Ion Channel can be Tuned by Mutation of Hydrophobic Residues in the Pore ¨ zge Yoluk1, Stephanie Heusser2, Iman Pouya1, Rebecca Howard3, O Go¨ran Klement2, Erik Lindahl1,2. 1 Theoretical & Computational Biophysics, KTH Royal Institute of Technology, Stockholm, Sweden, 2Biochemistry & Biophysics, Stockholm University, Stockholm, Sweden, 3Chemistry, Skidmore College, Saratoga Springs, Sweden. Ligand-gated ion channels exhibit a wide range of responses, and the very same allosteric modulator can either inhibit or potentiate channels depending on single-residue mutations. We have previous explained this with multiple binding sites where binding can have opposite effects, which would have farreaching implications for the design of new pharmaceuticals. However, we still know very little about exactly why channels open or close in response to agonists and allosteric modulators. Here, we have used extensive ensemble computer simulations to show that mutations of hydrophobic ILE to hydrophilic residues such as SER in the 9’ and 16’ locations of the central ion pore of ligand-gated ion channels will alter the hydration, which leads to a more wider pore region. By combining simulations with electrophysiology recordings we have been able to show that while some of these mutations increase currents, others have the opposite effect and will make the channel harder to open. In contrast, intermediate-hydrophobicity mutations to ALA have the opposite effect and facilitate channel opening. This is explained by the double ring of SER residues creating a binding site, which likely causes a monovalent ion block, similar to the behavior for divalent ions in the wild-type. This shows the opening/closing in GLIC is due to a delicate balance of hydrophobic and hydrophilic interactions in the pore, it supports that this balance can be altered by mutations, and it indicates the actual ion conductance is not simply correlated to the dimensions of the pore, but rather the absence of binding sites inside the pore combined with high levels of hydration. In particular, this makes it possible to tune ligand-gated channel response, and possibly the allosteric modulation. 2169-Pos Board B306 Glic-Elic Chimeras have Unexpected Characteristics Sarah C. Lummis, Mona Alqazzaz. Dept of Biochemistry, University of Cambridge, Cambridge, United Kingdom. The prokaryotic ligand-gated ion channels from Gloeobacter violaceus (GLIC) and Erwinia chrysanthemi (ELIC) have provided useful information about the homologous Cys-loop family of neurotransmitter receptors. GLIC is of particular interest for the study of gating transitions, in that its structure has been elucidated in multiple conformational states under a number of conditions. However its mechanism of action is controversial - the protonation of His11’ in M2 has been shown to be essential for the activation of GLIC(1,2), yet a GLIC-glycine chimera retains proton sensitivity without the GLIC M2 domain(3). Here we create an ELIC-GLIC and a GLIC-ELIC chimera, express them in Xenopus oocytes, and test them for function using TEVC. The ELICGLIC chimera was not activated by 100mM GABA or pH 4. The GLIC-ELIC chimera was not activated by GABA, but was activated by protons with a pH50 of 6.6, similar to the GLIC-glycine chimera but not WT GLIC (pH50¼5.5). We 432a Tuesday, February 10, 2015 then tested the GLIC-ELIC chimera with crotonic acid and picrotoxin. Crotonic acid inhibits GLIC with an IC50 of 110mM; our data indicate it binds to the extracellular domain. Picrotoxin (IC50¼2.6mM) blocks the GLIC pore(4); it likely cannot access the ELIC pore(5), but may bind to the extracellular domain (IC50¼96mM;6). These compounds were less potent than expected in the chimera (IC50>300mM). Overall the data suggest that domain specific effects may not be accurately reproduced in complex chimeras with intercommunicating domains, such as an orthosteric binding site and a pore in ligand-gated ion channels. 1. Rienzo et al. (2104) Chem.Biol. (in press); 2.Wang et al. (2012) J.Biol.Chem. 287: 6482; 3.Duret et al. (2011). PNAS 108: 12143; 4.Alqazzaz et al., (2011) Biophys.J. 101: 2912; 5.Ulens et al.(2014) Structure(in press). 6.Thompson et al. (2012) Neuropharm. 63:761. 2170-Pos Board B307 Role of the Transmembrane a-Helix M4 in the Potentiation of Pentameric Ligand-Gated Ion Channels Camille M. He´nault1, Casey L. Carswell1, Sruthi Murlidaran2, J.P. Daniel Therien1, Peter F. Juranka1, Julian A. Surujballi1, Grace Brannigan2,3, John E. Baenziger1. 1 Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, Canada, 2Center for Computational and Integrative Biology, Rutgers University-Camden, Camden, NJ, USA, 3Physics, Rutgers University-Camden, Camden, NJ, USA. The gating of pentameric ligand gated ion channels (pLGICs) is sensitive to a variety of allosteric modulators that act on the transmembrane domain, including lipids. Here, we use two prokaryotic homologs, GLIC and ELIC, to examine the role of the lipid-exposed transmembrane a-helix, M4, in the allosteric regulation of pLGICs. Aromatic interactions at the interface between M4 and the adjacent a-helices, M1 and M3, drive M4 binding during folding (Haeger et al. (2010) Nat Struct Mol Biol 17:90-98). Alanine substitutions of the aromatics at this interface in GLIC weaken M4 binding and inhibit channel function, while aromatic substitutions of aliphatic residues at the same interface in ELIC promote M4 binding and potentiate channel function. The strength of M4 binding to M1/M3 governs the susceptibility of both pLGICs to the potentiating effects of a congenital myasthenic syndrome mutation, which occurs in muscle-type nicotinic receptors on the lipid-facing surface of M4 and alters lipid-protein interactions to enhance function, but has little effect on the inhibitory activity of the drug, propofol. We thus identify both M4-dependent and M4-independent pathways for transmembrane domain allosteric regulation. Our data suggest that the chemistry at the interface between M4 and M1/M3 influences the intrinsic M4 binding affinity for M1/M3 and may thus govern the susceptibility of a pLGIC to the potentiating effects of lipids. 2171-Pos Board B308 The Kinetic Properties of the Human Glycine Receptor in Response to Different Agonists Elliot J. Hurdiss1, Timo Greiner1, Rilei Yu2, Phillip C. Biggin2, Lucia G. Sivilotti1. 1 Department of Neuroscience, Physiology and Pharmacology, UCL, London, United Kingdom, 2Structural Bioinformatics and Computational Biochemistry, Department of Biochemistry, Oxford University, Oxford, United Kingdom. Glycine receptors are ligand-gated chloride channels that mediate much of the fast synaptic inhibition in the spinal cord and brainstem. They can exist as homomers (formed by five a1 subunits), or as heteromers (formed by 3 a1 subunits and 2 b subunits). One of our current lines of work is to understand the effect on glycine receptors of a range of agonist molecules with structures systematically changed from that of glycine. In this study, we selected two agonists, b-alanine and D-alanine, for detailed single channel analysis. By fitting kinetic schemes to cell-attached data we were able to get insight into the binding and gating of the glycine receptor in response to the different agonists. We also used concentration jump experiments in the outside-out configuration to validate the fitted data. Molecular dynamics simulations also allowed us to see how the agonists docked into the binding site. While binding and gating rate constants remained relatively similar between all agonists, the ability of the agonists to stabilise the pre-open flipped confirmation varied in a manner similar to that reported by Lape et al. (Nature, 2008. 454 722-729). The less effective agonist D-alanine proved also to have a lower binding affinity than the other two agonists. Molecular docking studies were able to confirm that both D-alanine and b-alanine bind in a similar fashion to glycine. However, b-alanine shows a lot more mobility in the binding site, and this perhaps accounts for the large amount of heterogeneity in single channel recordings with this agonist. 2172-Pos Board B309 Evolution of Pro-Loop Channels: A Fresh Look at the Former Cys-Loop Family Mariama Jaiteh, Antoine Taly, Je´roˆme He´nin. IBPC, CNRS, Paris, France. Cys-loop neurotransmitter-gated ion channels were a well-known superfamily of synaptic receptors. A small revolution occurred when Tasneem et al. identified prokaryotic members of the family that lacked both the eponymous cysteines and obviously the neuronal context of their eukaryotic relatives. A decade later, a new foray into the phylogeny and evolution of the family brings more surprises still, including related channels in various unicellular eukaryotes, as well as Cys-less members in a number of Metazoan species. This prompts a significant rewrite of the evolutionary history of the more aptly named ‘‘Pro-loop’’ channels, while leaving many more questions open. 2173-Pos Board B310 Electromagnetic Fields Inhibit Cys-Loop Receptor Function by Inducing a Novel Conformational State Fernanda Tolosa1, Walter R. Cravero2, Cecilia B. Bouzat1. 1 Instituto de Investigaciones Bioquimicas, Bahia Blanca, Argentina, 2 Universidad Nacional del Sur, Instituto de Fisica del Sur, Bahia Blanca, Argentina. The fact that the general population is increasingly exposed to electromagnetic fields (EMFs) due to the advances in technology raises concern about their potential health effects. There are many gaps in knowledge, particularly at the molecular level, still needing to be filled before better health risk assessments can be made. We therefore studied the influence of EMFs on two Cys-loop receptors: muscle nicotinic (AChR) and 5-HT3A receptors. The transient exposure of cells expressing these receptors to EMFs (15 Hz-120 kHz) significantly decreases the peak current and increases the rise time of macroscopic currents elicited by the agonists. The peak current decreases as a function of EMF frequency (IC50¼ 54 kHz for AChR). The effects on both receptors are qualitatively similar but more profound for 5-HT3A, indicating different sensitivity to the EMF within the receptor family. To understand the molecular basis leading to the macroscopic changes, we compared single-channel properties before and after the exposure to EMFs. Singlechannel amplitude, open duration, duration of activation episodes (clusters) and open probability within clusters are not affected by the EMF. However, EMF leads to a profound decrease of the number of clusters as a direct function of frequency. The analysis reveals that EMFs induce a novel, non conductive, conformational state that arises from the closed resting state through a frequency-dependent transition. Thus, the stabilization of this novel state by EMF sequesters receptors from the activation pathway. Simulations of macroscopic and single-channel currents on the basis of a scheme including this new state well reproduce our experimental data. The identification of a novel conformational state induced by EMF enhances our understanding of receptor function, in general, and of the mechanisms by which EMFs affect neuronal excitability, in particular. 2174-Pos Board B311 Super-Resolution Imaging and Single Particle Tracking of Serotonin 5HT3A Receptor in Biomimetic Membranes Adam O. Barden, Adam S. Goler, James A. Brozik. Chemistry, Washington State University, Pullman, WA, USA. The serotonin 5HT3 receptor is a member of the Cys-loop super family of ligand gated ion channels (LGIC). Like all members of this family, 5HT3 is composed of five independent subunits. The receptor has a large extracellular domain, with the ion channel being constructed from the second transmembrane segment of each subunit, and there is a smaller intercellular region that is thought to interact strongly with cytoskeletal proteins. We have shown that the 5HT3 receptor can be efficiently incorporated into planar supported biomimetic membranes and the orientation can be controlled. The mobility of 5HT3 is greatly affected by both the orientation of the LGIC and the composition of the biomimetic assembly. We have used single molecule fluorescence imaging to track the mobility of individual proteins within the supported lipid bilayer and have used c-terminal labeling to determine the orientation of individual proteins. We have recently constructed a low temperature superresolution fluorescence microscope and have carried out experiments in frozen assemblies in order to identify the individual subunits that compose 5HT3A (the homopentamer of 5HT3). Tuesday, February 10, 2015 2175-Pos Board B312 Interaction of Bupropion with 5-HT3A Receptors Akash Pandhare1, Dominique Gagnon1, Henrik Wilms2, Michael P. Blanton3, Michaela Jansen1. 1 Cell Physiology and Molecular Biophysics, Texas Tech University Health Sciences Center, Lubbock, TX, USA, 2Neurology, Texas Tech University Health Sciences Center, Lubbock, TX, USA, 3Pharmacology and Neuroscience, Texas Tech University Health Sciences Center, Lubbock, TX, USA. For more than two decades, bupropion has been clinically prescribed for the treatment of depression (WellbutrinÒ), and more recently for smoking cessation (ZybanÒ). Bupropion is conventionally described as a dual norepinephrine dopamine reuptake inhibitor, therefore effective as an antidepressant. However, we and others have shown that it also interacts with receptors of the Cys-loop superfamily, namely neuronal nicotinic acetylcholine receptors. In the continuum of exploring the molecular interactions of bupropion with other prominent members of the Cys-loop superfamily, we report here that bupropion additionally inhibits 5-hydroxytryptamine (serotonin) type 3A receptors (5-HT3ARs). 5-HT3ARs are cation-conducting, homo-pentameric ligand-gated ion channels of the Cys-loop superfamily which are structurally and functionally distinct from other serotonin receptors that are G-proteins. 5-HT3 receptors are current targets for anti-emetics mainly used in cancer chemotherapy, as well as potential future targets for disorders including anxiety, schizophrenia, and Alzheimer’s disease. Here we characterized our newly-identified interaction of bupropion with 5HT3ARs with electrophysiological and radioligand binding studies. In oocytes, bupropion inhibited 5-HT3AR-mediated currents with an IC50 value of 87 mM (nH ¼ 1.2). That 300 mM bupropion inhibited [3H]-5-HT or [3H]-granisetron (a competitive antagonist) binding by ~ 20% indicated at best weak interaction with the agonist binding site. In contrast, the effect of different concentrations of bupropion on serotonin concentration-response curves suggested a noncompetitive nature of interaction. Along these lines, importantly, the inhibition of serotonin-evoked currents recorded from oocytes was proportional to the length of preincubation time with bupropion. In summary, our results are indicative of a negative allosteric interaction of bupropion with the 5-HT3AR. Currently, experiments are underway to identify the site(s) of interaction for bupropion within the 5- HT3AR. 2176-Pos Board B313 Exploring the Gating Pathway in an Eukaryotic Ligand-Gated Ion Channel Stephanie A. Heusser1,2, Ozge Yoluk1,3, Erik Lindahl1,3. 1 SciLifeLab, Solna, Sweden, 2Stockholm University, Stockholm, Sweden, 3 KTH, Stockholm, Sweden. Pentameric ligand-gate ion channels (pLGIC) such as the GABAA-, 5-HT3and Glycine receptor play a crucial role in neurotransmission and are targeted by a wide variety of drugs including anesthetics, benzodiazepines, and anticonvulsants. Despite their high pharmacological importance, little is known about the pathway from neurotransmitter binding in the extracellular domain to the opening of the channel in the transmembrane domain. Partly, this is due to the lack of high-resolution structural data of pharmacologically relevant receptors. Recently however, the crystallographic structures of the C. elagans glutamate-gated chloride channel (GluCl) both in an apo- as well as in a presumed open state became available (PDB: 4TNV, 4TNW; 3RHW). Based on theses structures along with observations made from molecular dynamics simulations, we focused our interest on one specific loop in the extracellular domain. We have used electrophysiological experiments on Xenopus oocytes to explore the effect of mutations in loop F on the activation of GluCl by glutamate. We also investigated the influence of the partial allosteric agonist ivermectin on glutamate response both in presence and absence of the same mutations in loop F. We found that mutation of a residue in this region has a significant influence on glutamate response. This suggests that this region plays an important role in the pathway between ligand binding to channel opening. These insights should help to better understand the underlying mechanism of this receptor family and might open up new possibilities for drug treatment. 2177-Pos Board B314 Complex Modulation of the GABAA a1b2g2 Receptor Function by Bupropion Jeremy M. Thompson1, Aneesh Pappu2, Akash Pandhare3, Michaela Jansen3. 1 Pharmacology and Neuroscience, Texas Tech University Health Sciences Center, Lubbock, TX, USA, 2Clark Scholars Program, Texas Tech University, Lubbock, TX, USA, 3Cell Physiology and Molecular Biophysics, Texas Tech University Health Sciences Center, Lubbock, TX, USA. 433a Bupropion is one of the most common drugs prescribed for treating depression and aiding smoking cessation, and is generally well tolerated with the exception of a rare dose-dependent incidence of seizures. It inhibits norepinephrine and dopamine reuptake, as well as noncompetitively antagonizes neuronal nicotinic acetylcholine receptors, which are members of the Cys-loop superfamily. While investigating its effects on other key members of this superfamily, bupropion showed complex modulation of the g-aminobutyric acid type A (GABAA) a1b2g2 receptor function. In the brain, GABAA a1b2g2 receptors are one of the most abundant chlorideconducting hetero-pentameric ligand-gated ion channels. They are targets for a number of drugs including anesthetics, anticonvulsants, anxiolytics, hypnotics, and muscle relaxants. To the best of our knowledge, the interaction of bupropion with the GABAA receptor has not yet been described. Therefore, the primary goal of this study was to characterize the functional interactions of bupropion with the GABAA a1b2g2 receptor. In oocytes, bupropion-alone (0.1 mM - 3 mM) directly activated GABAA receptor-mediated currents. The comparison of doseresponse curves showed that bupropion-activation was lower in potency (EC50¼1.5 mM vs. 1.3 mM) and efficacy (49.5% vs. 100%) than that for GABA, consistent with partial agonism of the GABAA receptor. Interestingly, at lower concentrations (0.001 mM - 100 mM) bupropion inhibited 0.5 mM GABA-induced currents by ~ 8 - 20%; in contrast, at higher concentrations (1 mM - 3 mM) it potentiated GABA-induced currents by ~ 50 - 70%. Notably, in many ways bupropion mimics the actions of some of the general anesthetics (e.g. propofol) at the GABAA receptor. This warrants exploring the bupropion binding site(s) within these receptors. 2178-Pos Board B315 Building GABAA Receptors for Structural Determination Duncan C. Laverty1, Adam Cryar2, Konstantinos Thalassinos2, Trevor G. Smart1. 1 Neuroscience, Physiology & Pharmacology, University College London, London, United Kingdom, 2Institute of Structural and Molecular Biology, University College London, London, United Kingdom. GABA is the predominant mediator of inhibitory neurotransmission in the brain, and upon release rapidly activates ionotropic GABAARs. These receptors are members of the pentameric ligand gated ion channel (pLGIC) family and are the targets of a range of clinical and endogenous allosteric modulators, including general anesthetics, alcohols and neurosteroids. Many of these compounds bind in the transmembrane domain (TMD) of GABAARs and selectively potentiate or inhibit activity, and in some cases directly gate the channel. A number of crystal structures of prokaryotic and eukaryotic pLGICs have now been solved, including the first GABAAR structure, the human b3 homopentamer. However, high resolution structures that reveal the molecular basis for allosteric modulation of GABAARs are currently unavailable. Here we report the construction, characterization and crystallization of a chimeric pLGIC, which is amenable to structural studies of GABAAR transmembrane pharmacology. We have generated a chimeric receptor of the extracellular segment from the proton-gated ion channel from Gloeobacter violaceus (GLIC) and the transmembrane segment of the anion-selective human a1 GABAA receptor. The GLIC-a1 chimera formed functional proton-gated channels when expressed in Xenopus oocytes. Proton gated currents of the chimeric channel were potentiated by positive modulatory neurosteroids and depressed by inhibitory neurosteroids, whilst GLIC is normally insensitive to these compounds. This receptor can be overexpressed in insect cells and used to generate purified and intact homopentameric receptor (as determined by native mass spectrometry). Furthermore the purified protein can be used to grow protein crystals under a range of conditions. We are currently carrying out crystallization trials to generate diffraction quality crystals of our chimera for structural determinations. 2179-Pos Board B316 Dopamine Directly Modulates GABAA Receptors Paul Hoerbelt, Mark W. Fleck. Albany Medical College, Albany, NY, USA. Dopamine is a critical neuromodulator that governs movement and motivation. Although dopamine is only thought to target GPCRs in vertebrates (D1-5), it can activate a Cys-loop pentameric ligand-gated ion channel in C. elegans (LGC-53). This channel shares homology with mammalian GABAA receptors, among other Cys-loop channels. The GABAA b3 subunit in particular is important for mediating tonic GABA signaling in dopamine-projecting striatal medium spiny neurons. We found 30% residue identity between ligand-binding domains of LGC-53 and b3, and hypothesized that b3-containing GABAA receptors may be directly modulated by DA. To address this, we used immunolabeling with specific anti-b3 antibodies and whole-cell patch clamp 434a Tuesday, February 10, 2015 electrophysiology in cultured rat striatal neurons and transfected HEK 293 cells. We determined that 79 53% of cultured cells endogenously expressed b3, and 94% of neurons gave tonic-level GABA-evoked currents blocked by phasic concentrations of dopamine (0.1-10 mM). Inhibition was recapitulated in HEK 293 cells transfected with a1b3, a1b3d or a1b2g2 subunits, but we instead observed potentiation in a1b3g2 and a5b3g2. Surprisingly, dopamine (1 mM) evoked rapid currents in a1b2g2, a1b3g2 and a5b3g2 in the absence of GABA. In a1b3(H267A)g2 (a1Zb3g2) receptors insensitive to trace Zn2þ inhibition, we found that other biogenic amines evoked comparatively smaller currents than DA. When the ratio of a1 was increased or we mutated a critical Zb3 tyrosine residue (Y62L), relative dopamine responses decreased. Furthermore, dopamine activity was retained but GABA activity was drastically reduced in Zb3g2 receptors, while Zb3 or g2 alone did not elicit currents. Finally, dopamine currents were picrotoxin- but not bicuculline- or gabazinesensitive. Taken together, dopamine at phasic levels is a GABAA allosteric modulator that may inhibit tonic GABA in prototypical a-containing GABAA receptors. However, at b/g-containing receptors that do not need a subunits, dopamine acts as a positive allosteric modulator. 2180-Pos Board B317 MmTX1 and MmTX2 from Coral Snake Venom Potently Modulate GABA(A) Receptor Activity Jean-Pierre Rosso1, Ju¨rgen R. Schwarz2, Marcelo Diaz-Bustamante3, Brigitte Ce´ard1, Jose´ Marı´a Gutie´rrez4, Matthias Kneussel2, Olaf Pongs5, Frank Bosmans6, Pierre E. Bougis1. 1 Aix Marseille Universite´, Marseille, France, 2University Medical Center Hamburg-Eppendorf, Hamburg, Germany, 3Lieber Institute for Brain Development, Johns Hopkins University, Baltimore, MD, USA, 4Universidad de Costa Rica, San Jose, Costa Rica, 5Universita¨t des Saarlandes, Homburg, Germany, 6Physiology, Johns Hopkins University, Baltimore, MD, USA. GABA(A) receptors shape synaptic transmission by modulating Cl- conductance across the cell membrane. Remarkably, animal toxins that target GABA(A) receptors have not yet been identified. Here, we report the discovery of MmTX1 and MmTX2, two toxins present in Costa Rican coral snake venom that bind to GABA(A) receptors at nanomolar concentrations. Studies with recombinant and synthetic toxin variants on hippocampal neurons and cells expressing common receptor compositions suggest that MmTX1 and MmTX2 potentiate GABA(A) receptor opening and accelerate desensitization when an agonist is present. In concert, hippocampal neuron excitability measurements reveal a toxin-induced transitory network inhibition followed by an increase in spontaneous activity. Moreover, toxin injections into mouse brain result in reduced basal activity between intense seizures. Altogether, we characterized the first animal toxins that potently modulate GABA(A) receptor function, thereby establishing a novel class of tools to study this receptor family. 2181-Pos Board B318 Monitoring Motions in GABA-A Receptor Intersubunit Transmembrane Cavities Tzu-Wei Tsao1, Borna Ghosh2, Cynthia Czajkowski1. 1 Department of Neuroscience, University of Wisconsin-Madison, Madison, WI, USA, 2Washington University at St. Louis, St. Louis, MO, USA. GABA-A receptors (GABAARs) are the target for many therapeutic drugs including benzodiazepines (BZDs), barbiturates, anesthetics and neurosteroids. These drugs bind to distinct sites and potentiate GABA-induced currents. They are often used in combination but little is known about mechanism(s) underlying their interactions. Recent crystal structures of GLIC and the glutamateactivated chloride channel suggest agonist-mediated channel opening is associated with expansion of transmembrane interfaces between M3 and M1 helices of adjacent subunits. Here, we examined if binding of different allosteric drugs and combinations of these drugs to a1b2g2L GABAARs induce similar motions. We also tested whether potentiation of GABA-mediated current by pairs of drugs binding to distinct sites are additive or super-additive. We individually inserted a cysteine in the M1 helices of each GABAAR subunit at a1I227, b2L223 and g2LI238 to monitor motions at different intersubunit interfaces. We expressed wild-type and mutant GABAARs in Xenopus oocytes. We measured rates of modification of the substituted cysteines in the absence and presence of different combinations of allosteric modulators that bind to different sites: the intravenous anesthetics pentobarbital (PB) and etomidate, the neurosteroid THDOC and the benzodiazepine flurazepam. When applied alone, flurazepam and THDOC induced distinct motions in the intersubunit interfaces, flurazepam increased rate of modification of only gI238C whereas THDOC only increased the rate of bL223C. When flurazepam and THDOC were co-applied, no change in rate of modification of gI238C was observed indicating that the presence of THDOC inhibited the ability of flurazepam to elicit structural rearrangements near gI238C. The data suggest structural mechanisms underlying the effects of combining drugs are different than mechanisms underlying their actions alone. Ion Channel Regulatory Mechanisms II 2182-Pos Board B319 Liberation of Pser68-Plm Inhibition of NCX1 by an Optimized Anchoring Disruptor Peptide Kjetil Hodne1,2, Pimthanya Wanichawan1,2, Tandekile Lubelwana Hafver1,2, William Edward Louch1,2, Marianne Lunde1,2, Marita Martinsen1,2, Ole Mathias Sejersted1,2, Cathrine Rein Carlson1,2. 1 Institute for Experimental Medical Research, Oslo University Hospital and University of Oslo, Oslo, Norway, 2KG Jebsen Cardiac Research Center and Center for Heart Failure Research, Oslo, Norway. The cardiac sodium-calcium exchanger (NCX1) is an important regulator of intracellular Ca2þ ([Ca2þ]i) and a potential therapeutic target in heart failure. Among potential interacting partners regulating NCX1 activity is the transmembrane protein phospholemman (PLM); a substrate for protein kinase A and protein kinase C. Several reports have demonstrated that binding of phosphorylated PLM (pSer68-PLM) to NCX1 leads to NCX1 inhibition, while other studies have failed to demonstrate a functional interaction of these proteins. In present study, we aimed to analyze the biological function of the pSer68-PLMNCX1 interaction by developing anchoring disruptor peptides. We observed that PLM co-fractionated and co-immunoprecipitated with NCX1 in rat left ventricle (LV) and in co-transfected HEK293 cells. Strong binding of pSer68-PLM to one of the previously reported NCX1-QKHPD regions was demonstrated in pull-down and overlay assays. Single amino acid substitutions of native NCX1 peptide sequence 1-KHPDKEIEQLIELANYQVLS-20 revealed that single substitutions at position 1-11 (particularly with tryptophan or tyrosine) increased binding affinity of the peptide to pSer68-PLM. Interestingly, we found that double substitution at position 4 and 6 with tyrosine (D4Y and E6Y) in the native peptide sequence enhanced the binding affinity to pSer68-PLM 8-fold, compared to native sequence. The optimized peptide 1KHPYKYIEQLIELANYQVLS-20 was further tested in a series of electrophysiological experiments using whole-cell voltage clamp technique, with voltage-ramp protocol employed to elicit NCX current. Constitutively phosphorylated (S68D) PLM was observed to inhibit NCX1 current in both forward and reverse mode. Inclusion of the disruptor peptide in the patch pipette reversed S68D-PLM inhibition of NCX1, while NCX current was not altered by a scrambled peptide control. Taken together these data strongly suggest that PLM interacts directly with NCX1, and that when PLM is phosphorylated at serine 68, the interaction inhibits NCX1 activity. 2183-Pos Board B320 The Talk-1 C-Terminus is a Charge-Sensitive Module Regulating Channel Activity Nicholas Vierra1, Prasanna K. Dadi1, Farah Ladak2, David A. Jacobson1. 1 Molecular Physiology & Biophysics, Vanderbilt University, Nashville, TN, USA, 2Eastern Virginia Medical School, Norfolk, VA, USA. Human beta-cell insulin secretion is dependent on Ca2þ entry through voltagedependent Ca2þ channels (VDCCs). Kþ channels modulate the beta-cell membrane potential, which influences VDCC activity. The most abundant Kþ channel of the human islet is the two-pore domain Kþ (K2P) channel TALK-1; however, its function is unknown. We characterized TALK-1 currents in primary mouse beta-cells and find that K2P currents are significantly reduced after genetic ablation of TALK-1. To identify mechanisms that regulate TALK-1 channel activity, we used a Tlþ-sensitive fluorescent screen to assess if TALK-1 is regulated by kinases. Cells expressing TALK-1 channels were monitored for changes in Tlþ flux in response to co-expression with a constitutively active kinase (examining a total of 192 kinases), revealing 9 kinases that modify Tlþ flux through TALK-1. Interestingly, three phosphatidylinositol phosphate kinases (PIKs) including PIK3CG, PI4K2B, and PIKFYVE, increased TALK-1 channel activity. To determine if activation of TALK-1 channels by PIKs is due to build-up of PIPs, we depleted membrane PIP2 with a rapamycin-recruitable phosphatase and found that TALK-1 channel activity was reduced. As charged residues of other Kþ channels mediate PIP2 sensitivity, we assessed if PIP2 influences TALK-1 channel activity through a charged residue enriched domain in the C-terminus. We found that neutralization of these charged residues significantly reduced PIP2 activation of TALK-1 channels. Importantly, PIP2 introduced via patch pipette into primary mouse beta-cells also significantly enhanced TALK-1 currents. As glucose induces Ca2þ-dependent oscillations in beta-cell PIP2, we then assessed how TALK-1 regulates islet Ca2þ dynamics. We find that Ca2þ oscillation frequency is significantly increased in islets lacking TALK-1, which Tuesday, February 10, 2015 results in enhanced glucose-stimulated insulin secretion (GSIS). These data provide compelling evidence that TALK-1 influences GSIS by limiting excitability, and suggests a novel mechanism for phospholipid-dependent regulation of beta-cell Ca2þ entry. 2184-Pos Board B321 Investigating Viral Channel Forming Protein VPU with Coarse-Graining Molecular Dynamics Simulation Meng-Han Lin, Wolfgang Fischer. Institute of Biophotonics, Taipei, Taiwan. VPU of HIV has been verified as a channel-forming protein by experiment. The self-assembly process and the oligomer state of VPU are not clear. We use coarse-graining molecular dynamics simulation to study the full-length channel-forming protein VPU in lipid bilayer. The full-length of VPU was constructed by linking the cytoplasm and transmembrane domain solved by NMR. We put different number of VPUs 2, 3, 4, 5 or bigger amount 16, 32 in simulation box. Our results show several binding sites and the oligomer state. In those simulations, the four VPUs form a symmetric bundle which is close to the channel structure. The tetramer was seem to form via a dimer. Not only transmembrane domain but also cytoplasm domain was involved in self-assembly. 2185-Pos Board B322 Kir2.1 Channels Compensate for the Loss of KATP Channels in SUR1 Null Islets Suryakiran Vadrevu1, Jinhua Ren1, Min Zhang2, Eric J. Glynn1, Arthur Sherman3, Leslie S. Satin1. 1 Pharmacology, University of Michigan, Ann Arbor, MI, USA, 2 Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, VA, USA, 3NIDDK, National Institutes of Health, Bethesda, MD, USA. Pancreatic islets of SUR1-/- mice exhibit electrical oscillations even though they lack KATP channels. However, while we confirmed that electrical oscillations persisted in freshly isolated SUR1-/- islets, after overnight culture in media containing 11.1 mM glucose, SUR1-/- islets exhibited continuous spiking in place of oscillations, as has been observed in human congenital hyperinsulinemia (CHI). Microarray analysis suggested upregulation of kcnj2, the gene encoding the inward rectifier Kir2.1, in SUR1-/- islets. To test whether Kir2.1 was upregulated in SUR1-/- islets islet lysates were immunoblotted for Kir2.1 protein. Kir2.1 was expressed in mouse and human islets and increased in lysates from freshly isolated SUR1 islets. Using patch clamping, we found that an inwardly rectifying K current was present in freshly isolated SUR1-/- islets that was absent in wild type islets. This current was sensitive to the Kir2.1 inhibitors ML133 and barium. Notably, rectifying current, enhanced Kir2.1 protein levels, and the expected Kir2.1 drug sensitivity were all lost along with oscillations after overnight culture in media with 11.1 mM glucose. Similar results were found using fura-2 loaded islets to examine Ca2þ oscillations. To test whether the suppression of Kir2.1 activity in cultured islets was a response to culture in high glucose, islets were cultured in 5 mM instead of 11.1 mM glucose. Lowering glucose preserved Kir2.1 protein and maintained the islet oscillations. These results show that the loss of KATP channels produced by deleting SUR1 is at least partially compensated for by the upregulation of Kir2.1 channels in mouse beta cells. We propose pharmacological activation of compensatory Kir2.1 channels as a novel way to inhibit the unrestrained insulin secretion characteristic of beta cells from human CHI patients. 2186-Pos Board B323 Secreted Human CLCA1 Activates Calcium-Dependent Chloride Currents via Interaction with TMEM16A (Anoctamin 1) Monica Sala-Rabanal1, Zeynep Yurtsever2, Colin G. Nichols1, Tom J. Brett3. 1 Cell Biology and Physiology, Washington University School of Medicine, Saint Louis, MO, USA, 2Internal Medicine, Washington University School of Medicine, Saint Louis, MO, USA, 3Biochemistry and Molecular Biophysics, Washington University School of Medicine, Saint Louis, MO, USA. The calcium-activated chloride channel (CaCC) regulator (CLCA) proteins were so named because their expression leads to generation of calciumdependent chloride currents (ICaCC) in mammalian cells; however, the molecular identity of the channel(s) mediating these currents, and the mechanisms through which CLCA1 regulates their activity, have remained unknown. In a previous study, we showed that human CLCA1 is a secreted self-cleaving zincin metalloprotease, and the resulting N-terminal fragment increases ICaCC density in HEK293T cells (J. Biol. Chem. 287, 42138-49, 2012). Here, we found that the same type of currents are activated in untransfected cells co-cultured with CLCA1-transfected cells or exposed to CLCA1-conditioned medium, indicating that secreted CLCA1 can activate ICaCC in a paracrine fashion. 435a Because CLCA1 and the CaCC TMEM16A (Anoctamin 1) are expressed in the same tissues, such as the airway epithelia, and are upregulated in the same pathologic states, such as asthma and COPD, we tested the hypothesis that CLCA1-modulated currents are carried by TMEM16A. We observed that CLCA1-induced whole-cell ICaCC in HEK293T cells overexpressing the protein is blocked in the presence of TMEM16A-specific inhibitors T16Ainh-A01 and N-((4-methoxy)-2-naphthyl)-5-nitroanhtranilic acid, and silenced in the presence of TMEM16A siRNA. By means of live cell flow cytometry assays using a novel fluorescent CLCA probe, we demonstrate that CLCA1 and TMEM16A physically interact at the cell surface, and our real-time qPCR, immunohistochemistry and confocal microscopy data suggest that CLCA1 drives TMEM16A surface expression. These results identify CLCA1 as the first secreted direct mediator of TMEM16A activity, and suggest that CLCA1 and TMEM16A operate together to generate ICaCC in multiple tissues. 2187-Pos Board B324 Inhibition of the CFTR Chloride Channel by Sphingomyelinase Brandon Stauffer, Guiying Cui, Daniel T. Infield, Nael McCarty. Emory University, Atlanta, GA, USA. CFTR is an epithelial chloride channel that controls hydration of multiple epithelia. It is well established that CFTR is regulated by phosphorylation of its Regulatory (R) domain and binding of ATP to its nucleotide binding domains (NDBs). In addition, evidence exists that sphingolipid metabolism may directly modulate CFTR activity. Specifically, it was shown that catalysis of sphingomyelin (SM) by sphingomyelinase (SMase) inhibits CFTR in Xenopus laevis oocytes. The mechanism by which the generation of ceramide inhibits CFTR is not known and the goal of this study is to elucidate the events leading to inhibition of CFTR chloride channel function following SMasemediated catalysis of SM. We set out to test whether common ceramidemediated events were responsible for CFTR inhibition. We first tested whether CFTR inhibition was the result of internalization by measuring the pH-sensitivity of fluorescence of externally-tagged GFP-CFTR and found that SMase treatment did not lead to massive internalization of CFTR. To determine whether CFTR inhibition resulted from alteration of ceramide-sensitive scaffolding proteins, for example ERM proteins, we tested whether a CFTR construct lacking a PDZ domain (DPDZ-CFTR) is sensitive to SMasemediated inhibition. Indeed, currents in DPDZ-CFTR expressing oocytes with purified SMase resulted in a disappearance of chloride current. Interestingly, we found that SMase did not inhibit CFTR currents in excised insideout patches. Taken together, these data suggest that SMase inhibits CFTR at the cell surface in a PDZ-independent manner and requires cytosolic signaling components. Support provided by NIH5R01DK075016. 2188-Pos Board B325 Molecular Dynamics Simulations of Calcium Binding Sites in the RCK Domain of the Mthk Gating Ring Tangzhen Zhao1, Yukun Wang2, Qin Xu2, Dongqing Wei2. 1 Physics and Astronomy, Shanghai Jiao Tong University, Shanghai, China, 2 Shanghai Jiao Tong University, Shanghai, China. Gating mechanism is one of the most important problems in the study on ion channels. Despite the important achievement of the high-resolution structures of several Kþ channels, the gating mechanism underlie the conformational change is still unclear. Here we employ the all-atom simulation method to study the gating ring of Mthk, a Ca2þ-activated Kþ channel, with a long time of 2 ms. we find two kinds of important Ca2þ binding sites. One plays a significant role in the stabilization of RCK structure; another one may participate in the conformational change of the gating ring structure and have a direct relation with the mechanism of gating. Morever, two arginines R132, R135 nearby the Ca2þ binding site E133, E258,E259 play a key role the process of conformational change. 2189-Pos Board B326 Functional Coupling between ANO1 and TRPV1 Channels in Sensory Neurons Shihab Shah, Nikita Gamper. Faculty of Biological Sciences, University of Leeds, Leeds, United Kingdom. Ca2þ activated Cl- channel ANO1 (TMEM16A) is one of the most recently discovered pro-algesic ion channels expressed in peripheral sensory neurons. In these neurons ANO1 is specifically activated by Ca2þ release from intracellular stores in response to inflammatory mediators such as bradykinin (BK). In addition, ANO1 is also activated by noxious heat in the temperature range that is similar to that of the heat sensor TRPV1. As TRPV1 is permeable to Ca2þ, there is an intriguing possibility that both channels can function in a coordinated fashion thus increasing the dynamic range of response to noxious heat. In order to investigate activation of ANO1 in nociceptors by intracellular 436a Tuesday, February 10, 2015 Ca2þ signals we developed a single-cell imaging approach that allows simultaneous monitoring of Cl- channel activity and intracellular Ca2þ concentration in cultured dorsal root ganglion (DRG) neurons using simultaneous imaging of halide-sensitive H148Q/I152L EYFP mutant and fura-2. We investigated activation of Ca2þ-activated anion conductance in DRG neurons in response to i) Ca2þ release from the IP3-sensitive intracellular stores induced by BK; ii) Ca2þ influx through the TRPV1 channels activated by capsaicin (CAP) and iii) Ca2þ influx via the voltage-gated Ca2þ channels (VGCC) induced by depolarization with extracellular solution containing 50 mM KCl (HK). Both BK and CAP produced large Ca2þ transients and caused significant quenching of YFP fluorescence. The onset of the YFP response to CAP was delayed as compared to the response to BK, possibly reflecting a shallower Ca2þ transient. Consistent with previous finding, activation of VGCC was least efficacious in activating CaCC and in approximately two thirds of neurons the YFP quenching was indistinguishable from the baseline rundown even despite strong Ca2þ transients produced by HK. Our results suggest that ANO1 in DRG neurons is preferentially coupled to colocalised Ca2þ sources. 2190-Pos Board B327 Phospholipase D2 Specifically Regulates TREK Channels via Direct Interaction and Local Production of Phosphatidic Acid Yannick Comoglio1, Levitz Joshua2, Michael Kienzler2, Florian Lesage3, Ehud Isacoff2, Guillaume Sandoz1. 1 iBV, CNRS, Nice, France, 2Department of Molecular and Cell Biology and Helen Wills Neuroscience Institute, UC Berkeley, Berkeley, CA, USA, 3 IPMC, CNRS, valbonne, France. Membrane lipids serve as second messengers and docking sites for proteins and play central roles in cell signalling. A major question about lipid signaling is whether diffusible lipids can selectively target specific proteins. One family of lipid-regulated membrane proteins is the TREK subfamily of K2P channels: TREK1, TREK2, and TRAAK. We investigated the regulation of TREK channels by phosphatidic acid (PA), which is generated by Phospholipase D (PLD) via hydrolysis of phosphatidylcholine. We found that, even though all three of the channels are sensitive to PA, only TREK1 and TREK2 are potentiated by PLD2 and that none of these channels is modulated by PLD1, indicating surprising selectivity. We find that PLD2, but not PLD1, directly binds to the C-terminus of TREK1 and TREK2, but not to TRAAK. The results lead to a model for selective lipid regulation by localization of phospholipid enzymes to specific effector proteins. Finally, by using the photoswitchable conditionnal subunit method to endow light sensitivity to the native TREK1 channels, we show that regulation of TREK channels by PLD2 occurs natively in hippocampal neurons. 2191-Pos Board B328 The Modulatory Function of the BK Channel g1 Subunit is Determined by its Transmembrane Domain Qin Li, Jiyuan Zhang, Karren Yen, Jiusheng Yan. MD Anderson Cancer Center, Houston, TX, USA. BK channels consist of pore-forming, voltage- and Ca2þ-sensing a-subunits (BKa), either alone or together with tissue-specific auxiliary b-subunits (b1-b4) or g-subunits. The newly identified g-subunits are a group of leucinerich repeat (LRR)-containing membrane proteins which contain a single transmembrane (TM) segment, a short intracellular C-terminal tail (C-tail), an N-terminal signal peptide, and a relatively large extracellular LRR domain. The g1 subunit (LRRC26), so far the most potent activator of BK channels, shifts the channel’s voltage dependence of activation in the hyperpolarizing direction by ~140 mV. We investigated the role and mechanism of the g1 TM domain in BK channel activation. We identified key amino acid residues in the g1 hydrophobic TM region involved in BK channel activation. We found that a minimum of 3 positively-charged residues on the intracellular sides of TM segment are also required to maintain the g1 subunit’s full modulatory function, which likely act through stabilization of the TM domain for proper association with BK channels. We also found that the single TM segment and its capping charged residues can fully retain the g1 subunit’s BK channel-activating efficacy. We conclude that the TM domain is a major contributor to the potent channel-activating efficacy of the BK channel g1 subunit. 2192-Pos Board B329 Differential Effects of PIP2 on Slo1 BK Channels with Different Auxiliary Subunits Yutao Tian1, Florian Ullrich2, Rong Xu1, Heinemann H. Stefan2, Shangwei Hou3, Toshinori Hoshi1. 1 Physiology, University of Pennsylvania, Philadelphia, PA, USA, 2 Biophysics, Friedrich Schiller University Jena & Jena University Hospital, Jena, Germany, 3Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University, Shanghai, China. Phosphatidylinositol 4, 5-bisphosphate (PIP2) regulates numerous ion channels, including large-conductance Ca2þ- and voltage-dependent Kþ (Slo1 BK) channels (Vaithianathan et al. 2008, J Gen Physiol 132:13-28). We examined the molecular and biophysical mechanisms of the effects of PIP2 on human Slo1 BK channels with different auxiliary subunits heterologously expressed in HEK cells. In the absence of heterologously expressed auxiliary subunits, bovine brain PIP2 (10 mM) applied to the cytoplasmic side inhibits currents through Slo1 channels. This inhibition is associated with a shift in GV by 18 mV and deceleration of the activation kinetics at positive voltages. In contrast, PIP2 markedly increases currents through Slo1þb1 and hSlo1þb4 channels by shifting their GV curves by 46 mV and 34 mV, respectively. The stimulatory effect of PIP2 on Slo1þb1 channels does not require Ca2þsensor activation or voltage-sensor activation. Currents through Slo1þb2 channels with a deletion in the N terminus of b2 to remove inactivation (D2-19) are not enhanced by PIP2 but neutralization of 3 negatively charged residues in the b2 D2-19 N terminus introduces a modest effect of PIP2. In Slo1þb1/b4, PIP2 accelerates the macroscopic activation kinetics at positive voltages and decelerates the macroscopic deactivation kinetics at negative voltages, but in Slo1þb2 D2-32 PIP2 has no effect on the deactivation kinetics. Measurements using chimeric b1-b2 subunits show that the second transmembrane domain and the C terminus of b1 is important for the large electrophysiological changes in Slo1þb1 channels caused by PIP2. Furthermore, Slo1 329RKK331, b1 R11, and b1 T14 are also critical to confer the effects of PIP2. Supported in part by the NIH, DFG HE 2993/8, and Shanghai Science and Technology Commission. 2193-Pos Board B330 A Fluorogen-Activating Biosensor for Analysis of BK Channel Traffic and Surface Residency Christopher Pratt1, Jianjun He2, Alison Barth1, Marcel Bruchez3. 1 Biological Sciences, Carnegie Mellon University, Pittsburgh, PA, USA, 2 Chemistry, Carnegie Mellon University, Pittsburgh, PA, USA, 3Biological Sciences, Chemistry, Carnegie Mellon University, Pittsburgh, PA, USA. The regulation of ion channels is critical to numerous physiological processes, especially neuronal function. The large conductance voltage and calciumactivated potassium (BK) channel can modulate neuronal excitability, neurotransmitter release, and may be involved in the development of epilepsy; following a seizure, hippocampal BK channels are more highly trafficked to the plasma membrane. In addition to the pore-forming alpha subunits, which are sufficient to confer channel function, Beta subunits can be incorporated to alter gating and trafficking. Studies of the brain-specific Beta4 subunit effects on surface expression have produced conflicting results, likely due to alternative splicing in the alpha subunit. Fluorogen-activating peptides (FAPs) developed in our lab are well-suited to study protein trafficking; consisting of a ScFv-derived fusion tag and fluorogen dyes added to the cellular media, the association of these two cognate parts results in bright, specific fluorescence. By using a FAP-tagged BKa construct and a pair of fluorogen dyes, we have developed a method for two-color labeling of surface-resident and intracellular BK channels in live cells, allowing for rapid analysis of protein localization by flow cytometry and direct imaging. Using this system in stably expressing HEK293 cells, we found that modulation of the C-terminal region by kinase activity exerted strong changes in surface expression. Taken together with previous observations that these same kinases alter channel gating, this provides a synergistic model for BK-mediated hyperexcitability. A common confound in BK channel research is due to the vast number of splicing isoforms, each having different sensitivities to post-translational modifications. To that end, we are currently generating a knock-in mouse model in which our FAPtagged BKa is included in all splice variants to examine trafficking changes in Vivo as well as analysis of the behavior of single channels. 2194-Pos Board B331 Understanding the Dynamics of K2P Channels in Complex Lipid Bilayers Prafulla Aryal1,2, Stephen J. Tucker2,3, Mark S.P. Sansom1,2. 1 Department of Biochemistry, University of Oxford, Oxford, United Kingdom, 2OXION Initiative in Ion Channels and Disease, University of Oxford, Oxford, United Kingdom, 3Department of Physics, University of Oxford, Oxford, United Kingdom. Ion channels are proteins that reside in phospholipid bilayers and conduct ions through their trans-membrane pore. Studying the dynamics of interaction between the channel, phospholipids, water and ions is fundamental to understanding ion channel function. We are using a combination of molecular dynamics simulations and functional tools to gain insight into K2P (Two Pore-domain Potassium Channel) function. By initially observing wetting and dewetting transitions in the inner pore of TWIK-1 (K2P1) channels during MD simulations and by subsequently combining in silico and in vitro mutagenesis and Tuesday, February 10, 2015 electrophysiology, we have found a hydrophobic barrier deep within the inner pore of the TWIK-1 channel [1]. Our study suggests that this barrier contributes to the very low level of functional currents observed for TWIK-1 channels. We have also reviewed the computational, structural and functional evidence for hydrophobic gating in several ion channel families and propose that understanding the dynamic behavior of water and ions within the pore represents an increasingly important element in understanding the relationship between ion channel structure and function [2]. We are now examining the interaction between K2P channels and phospholipids in more detail. Using MD simulations, we find hot spots for K2P channel and lipid interactions. These findings suggest that lipids can play modulatory roles in K2P channel function. [1] Aryal P, Abd-Wahab F, Bucci G, Sansom MSP & Tucker SJ. A hydrophobic barrier deep within the inner pore of the TWIK-1 K2P potassium channel. Nature Communications 5:4377 (2014) [http://dx.doi.org/10.1038/ ncomms5377] [2] Aryal P, Sansom MSP and Tucker SJ. Hydrophobic Gating in Ion Channels. Journal of Molecular Biology (2014) [http://dx.doi.org/10.1016/ j.jmb.2014.07.030] 2195-Pos Board B332 Non-Markovian Protein Dynamics in a Near-Critical Membrane Model Ofer Kimchi1, Benjamin B. Machta2. 1 Physics, Princeton University, Princeton, NJ, USA, 2Lewis-Sigler, Princeton University, Princeton, NJ, USA. Recent experiments in Giant Plasma Membrane Vesicles isolated from living cells have suggested that cell membranes are tuned close to a liquid-liquid critical point. Perturbations which influence membrane criticality - anesthetics and cholesterol level modulators - also affect the functioning of a large number of membrane-bound receptors and ion channels. This motivates us to develop a model for a membrane-bound protein that is allosterically regulated by interactions with its surrounding near-critical membrane. We consider a two dimensional lattice where Ising spins represent membrane lipids, and a two-state protein is represented as a group of like spins that must flip together. In our model, the full state of the system, including both protein and membrane degrees of freedom, obeys Markovian dynamics. However, when the protein is considered in isolation, as is typical experimentally, its dynamics become non-Markovian. We show that this phenomenon arises as information about the past state of the protein is stored in membrane degrees of freedom. Our model suggests a unified mechanism underlying the susceptibility of various ion channels to both anesthetics and cholesterol modulation and presents a new role for membrane lipids in the collective allosteric regulation of proteins. 2196-Pos Board B333 Mechanisms of TREK-2 Potassium Channel Gating Conor McClenaghan1, Elizabeth Carpenter2, Tucker J. Stephen1. 1 Department of Physics, University of Oxford, Oxford, United Kingdom, 2 Structural Genomics Consortium, University of Oxford, Oxford, United Kingdom. The K2P channel TREK-2 is an archetypal polymodal potassium channel which acts to couple a diverse range of regulatory stimuli to cellular electrical excitability. Alongside the other thermo-and mechano-gated K2P channels (TREK-1 and TRAAK) the TREK-2 channel is critical for discrimination of innocuous and noxious temperature and touch sensation. Guided by novel Xray crystal structures, we have used a variety of electrophysiological, pharmacological and kinetic studies to demonstrate a mechanism of action for the state-dependent inhibition of TREK-2 by norfluoxetine, a biologically active metabolite of the anti-depressant Prozac. These studies also enable us to propose a structural basis for activation of TREK-2 by membrane stretch, temperature and arachidonic acid. 2197-Pos Board B334 The Molecular Basis for Heme Modulation of KATP Channels Mark Burton1, Sofia Kapetanaki2, Noel Davies1, John Mitcheson1, Ralf Schmid3, Peter Moody3, Emma Raven2, Nina Storey1. 1 Cell Physiology and Pharmacology, University of Leicester, Leicester, United Kingdom, 2Chemistry, University of Leicester, Leicester, United Kingdom, 3Biochemistry, University of Leicester, Leicester, United Kingdom. Heme, iron protoporphyrin IX, is a vital prosthetic component of a number of functional hemoproteins playing essential roles in a diverse range of biological actions including oxygen transport, electron transport, catalysis and gene regulation. An emerging role of heme is its ability to regulate the activity of ion channels including voltage gated Kþ (Kv), large conductance Ca2þ-activated Kþ (BK) and epithelial Naþ (ENaC) channels. Here we report heme regulation of the ATP-sensitive Kþ (KATP) channel in both cardiac myocytes and 437a HEK293 cells heterologously expressing Kir6.2 and SUR2A subunits. The KATP channel is sensitive to the intracellular nucleotides ATP and ADP. The KATP channel links cellular metabolic state and excitability most notably during ischaemic stress. ATP acts with high affinity upon the cytosolic face to inhibit opening, thus the channel opens during periods of depleted cellular energy. Whole-cell KATP currents of both ventricular myocytes and HEK293 cells expressing KATP channels were increased upon application of 500 nM hemin extracellularly. In inside-out patches, KATP channel activity was reduced in the presence of ATP (500 mM), but on subsequent addition of hemin (500 nM) KATP channel open probability significantly increased from 0.024 5 0.013 to 0.110 5 0.028 (n ¼ 6, P < 0.01). Sequence alignments with known heme binding regions revealed a structurally unresolved region located on an intracellular linker in the non-pore forming SUR2A subunit containing the residues C628XXH631 and H648, which was analogous to the reported Kv1.4 heme binding sequence. Mutating these residues (C628S, H631A and H648A) led to reduced sensitivity of the resulting KATP channels to heme. Mutagenesis of each residue revealed C628 and H648 to have the greatest effect at reducing the agonistic effects of heme. Here we provide evidence for heme binding and regulation of KATP 2198-Pos Board B335 NMR Structural Studies of the Binding of Activating Mamba Toxin Tx7335 on the Potassium Channel KcsA Jing Zhu, Ulfat Shahzad, Sebastien F. Poget. Chemistry, College of Staten Island, Staten Island, NY, USA. We have recently identified a novel 63 amino acid residue three-finger toxin (called Tx7335) from eastern green mamba snake (Dendroaspis angusticeps) which interacts with KcsA and induces an increase in frequency and duration of individual channel openings when added to the outside of the channel. The toxin exerts this activating effect both on wild-type KcsA as well as on an agitoxin2-sensitive mutant form of the channel, indicating a mode of action and binding site that are different from the classic pore-blocker toxins. We are currently using NMR spectroscopy to unravel the structural underpinnings of this mechanism of action. High yield of purified 15N labeled KcsA and excellent NMR spectral quality have been achieved. Currently the characterization of toxin binding using 1H15N correlation spectra of 15N labeled KcsA in the absence and presence of toxin is ongoing. Experiments are conducted in different membrane mimetics including DMPC/DHPC bicelles and DPC or DM micelles at different pH, temperature and salt concentration. Some chemical shifts and peak intensity changes upon toxin addition have been observed. Continuing NMR structural studies will further elucidate the mechanism of how Tx7335 interacts with KcsA and shed light on the conformational and dynamic changes of C-type inactivation in KcsA and on a novel mechanism of ion channel regulation. 2199-Pos Board B336 Mechanism of Inhibition of the GluA1 AMPA Receptor Channel Opening by 2,3-Benzodiazepine Compounds Andrew Wu. Chemistry, SUNY at Albany, Albany, NY, USA. 2,3-Benzodiazepine derivatives, also known as GYKI compounds, represent a group of the most promising synthetic inhibitors of AMPA receptors. Here we investigate the mechanism of inhibition of the GluA1 channel opening and the site of inhibition by GYKI 52466 and its N-3 methyl-carbamoyl derivative (BDZ-f ) as well as two N-3 thiadiazolyl compounds. Like GluA2, GluA1 is a key AMPA receptor subunit, and excessive activity of GluA1 has been implicated in a number of neurological disorders. Using a laser-pulse photolysis technique, we investigated the mechanism of inhibition of the GluA1 channel expressed in HEK-293 cells. We found that these compounds inhibit the GluA1 channel noncompetitively. Addition of a methyl-carbamoyl group or a thiadiazole moiety to the N-3 position of the diazepine ring with the azomethine feature improves the potency of the resulting compounds without changing the site of binding, which we termed as the ‘‘M’’ site. On the basis of the magnitude of the inhibition constants for the same inhibitors (i.e., GYKI 52466 and BDZ-f), the ‘‘M’’ sites on GluA1 and GuA2 are different. Overall, the ‘‘M’’ site on GluA2 accommodates the same compounds better, or the same inhibitors show stronger potency on GluA2 than GluA1, if the N-3 pocket is not fully occupied. Acylating the N-3 position to occupy the N-3 side pocket of the ‘‘M’’ site can significantly narrow the difference and improve the potency of a resulting compound on GluA1. The two thiadiazolyl benzodiazepines inhibit both GluA1 and GluA2 much more strongly and almost equally potently. A thiadiazole moiety is thought to occupy more fully the side pocket of the ‘‘M’’ site, thereby generating a stronger, multivalent interaction between the inhibitor and the receptor binding site. This work is supported by NIH/NINDS. 438a Tuesday, February 10, 2015 2200-Pos Board B337 Simultaneous Measurements of Intracellular [Ca2D]i and [cAMP]i in Intact Islets to Study the Mechanism Underlying Dopaminergic Inhibition of Insulin Secretion Alessandro Ustione, David W. Piston. Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN, USA. Pancreatic islets secrete multiple hormones, including insulin, that are required to maintain euglycemia while meeting the energy demand of the body during everyday activities. We are interested in understanding the interplay between molecular mechanisms that precisely regulate these secretions. One specific paracrine modulator, dopamine, functions as a negative regulator of insulin secretion in the context of the pancreatic islet. It is secreted by the insulinproducing b-cells, activates an autocrine negative feedback that decreases the frequency of glucose-stimulated [Ca2þ]i oscillations, and in turn, inhibits insulin secretion. The G-protein coupled dopamine receptors are present in islet cells, but it is not clear how activation of the these receptors results in the observed changes in [Ca2þ]i in intact pancreatic islet cells. We are using an mTurquoise-Based cAMP biosensor with an improved dynamic range (Klarenbeek, J.B., et al., PLoS One, 2011), along with organic and genetically encoded Ca2þ-indicator dyes. Labeled cells are studied by live imaging using perfused pancreatic islet. Spectral unmixing is used to extract the fluorescence emissions. This experimental setup allows us to monitor the effect of specific dopamine receptor agonists and antagonists on the two main cellular second messengers. Also, islets from mice with a genetic target mutation of the DRD3 (D3-KO) are used to measure how the deletion of the dopaminergic feedback changes the second messenger dynamics. With the same approach we are measuring the effects of the overexpression of dopamine receptor D3 in wild-type and D3-KO islets. The information from these experiments will help elucidate the mechanism of dopamine signaling in the pancreatic islet. 2201-Pos Board B338 Determining the Dopaminergic Feedback Pathway in Pancreatic b-Cells with Fluorescence Fluctuation Spectroscopy Brittany Caldwell1, Alessandro Ustione2, David Piston1,2. 1 Biomedical Engineering, Vanderbilt University, Nashville, TN, USA, 2 Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN, USA. Tight regulation of insulin and glucagon, two hormones secreted by the pancreatic islet, allows the body to maintain glucose homeostasis. Insulin resistance coupled with insufficient insulin secretion leads to type 2 diabetes. Insulin secretion from pancreatic b-cells is regulated by multiple signaling inputs including the neurotransmitter dopamine. Upon treatment with dopamine, the amplitude and frequency of intracellular free calcium oscillations in b-cells decrease, leading to a concomitant decrease in insulin section. We hypothesize that activation of the dopamine receptor (DRD3) by dopamine releases the Gbg complex to stimulate a G protein-coupled inwardly-rectifying potassium channel (GIRK3). This inward current keeps the cell polarized, and reduces currents through voltage-gated calcium channels (CaV1.2). This model predicts numerous protein interactions that may only be transient in nature, in particular, we expect close interactions between the DRD3 and GIRK3 as well as each of these with the Gbg subunit. To test this hypothesis, we are utilizing fluorescence fluctuation spectroscopy to determine protein interactions in stable b-cell lines. Fluorescence fluctuation spectroscopy provides a method to determine interaction between two proteins in a live cell that surpasses many of the limitations of FRET. Single and two color studies allow us to determine diffusion constants of each protein before and after dopamine stimulation and the crosscorrelation, or amount of interaction, between two proteins. Additionally, brightness analysis can be used to determine homo- and heterodimerization of the tagged proteins. Diffusion constants, cross-correlation rates, and protein clustering among the DRD3, GIRK3, and Gbg subunit will be presented. Through this analysis, the dopamine signaling pathway in pancreatic b-cells can be determined, which will reveal novel potential therapeutic targets that can increase insulin secretion during diabetes mellitus. Other Channels 2202-Pos Board B339 Statistical Analysis of Multichannel Signals Rishabh Kumar1, Prashant Srinivasa2, Horia I. Petrache1. 1 Physics, Indiana University Purdue University Indianapolis, Indianapolis, IN, USA, 2Physics, California Polytechnic State University, San Luis Obispo, CA, USA. As opposed to single channel statistics, the analysis of superimposed signals is complicated due to signal overlap and loss of channel identity. We present a statistical method that uses a newly defined concept of multichannel events. These events can be easily differentiated and analyzed to provide information on single channel parameters such as ON and OFF times. 2203-Pos Board B340 Novel Step Detection Algorithms for Photobleaching Analysis of Protein Complexes with Many Subunits Nathan C. Deffenbaugh1, Yalei Chen1,2, Charles T. Anderson2,3, William O. Hancock1,2. 1 Department of Biomedical Engineering, Penn State University, University Park, PA, USA, 2Cell and Developmental Biology, Huck Institutes of the Life Sciences, Penn State University, University Park, PA, USA, 3Department of Biology, Penn State University, University Park, PA, USA. Single molecule photobleaching of fluorescently labeled protein complexes is an effective technique for counting constituent subunits and has been applied to determine the stoichiometry and oligomerization of several different transmembrane proteins including voltage- and ligand-gated ion channels, as well as the makeup of the cellulose synthesis complex. Fluorophore bleaching occurs as a random process, resulting in discrete intensity drops over time. Step detection algorithms can be used to identify these steps within noisy signals in order to determine the total number of fluorescent subunits within a protein complex. Reliable identification of steps can become difficult however, when high numbers of subunits are present. Here, we present two step detection algorithms, one based on modified t-testing and another based on the Bayesian Information Criterion (BIC), that perform at higher levels of precision than existing algorithms and account for temporal changes in variance within a signal, which is expected in photobleaching experiments. We also present a method for determining a unitary step amplitude associated with bleaching of a single fluorophore based on a Gaussian Mixture Model. 2204-Pos Board B341 Single-File Water Permeation through Aquaporin Channels Peter H. Nelson. Physics and Biology, Benedictine University, Lisle, IL, USA. A diffusive model of osmosis is presented that explains currently available experimental data. It makes predictions that distinguish it from the traditional convective flow model of osmosis, some of which have already been confirmed experimentally and others have yet to be tested. It also provides a simple kinetic explanation of Raoult’s law and the colligative properties of dilute aqueous solutions. The diffusive model explains that when a water molecule jumps from low to high osmolarity at equilibrium, the free energy change is zero because the work done pressurizing the water molecule is balanced by the entropy of mixing. It also explains that equal chemical potentials are required for particle exchange equilibrium in analogy with the familiar requirement of equal temperatures at thermal equilibrium. These are topics that should be considered for inclusion in the redesign of introductory physics courses for the life sciences (IPLS). The diffusive model also makes detailed predictions for the unidirectional fluxes through single-file aquaporins that can be tested experimentally or via molecular dynamics simulation. Predictions are made for both nonequilibrium and equilibrium simulations in which there may or may not be a water chemical potential difference across the membrane. The effects of both osmolarity and hydrodynamic pressure differences are included in the model. DUE-0836833 http://circle4.com/biophysics 2205-Pos Board B342 Making an Aquaporin Water-Tight: Structural Basis of Selectivity in Plant Nodulin 26 Intrinsic Proteins Zachary G. Beamer1, Tian Li2, Jerome Baudry1, Daniel M. Roberts1. 1 Biochemistry and Cellular and Molecular Biology, The University of Tennessee, Knoxville, TN, USA, 2Genome Science and Technology, The University of Tennessee, Knoxville, TN, USA. The evolution of land plants led toan amplification and diversification of the aquaporin superfamily of membrane channels. Among the subfamilies of plant specific aquaporin-like changes are the nodulin-26 intrinsic proteins (NIPs) which are multifunctional transporters of water, ammonia, glycerol and metalloid nutrients that participate in a number of osmoregulatory and metabolic functions. NIPs share the canonical hourglass fold of the aquaporin family, but possess substitutions within the aromatic arginine (ar/R) selectivity filter. The nine proteins of the NIP subfamily in the model plant Arabidopsis thaliana can be subdivided into two ar/R pore subgroups: the NIP subgroup I, which form aquaglyceroporins that are permeated by glycerol, water and ammonia, and the NIP subgroup II, which form metalloid transporters which are lack aquaporin activity and are essentially ‘‘water tight’’. These two NIP pore families differ principally by the substitution of a conserved alanine (NIP subgroup II) for a conserved tryptophan (NIP subgroup I) in the helix 2 position (H2) of Tuesday, February 10, 2015 the ar/R filter. Based on transport analyses and molecular dynamics simulation, a model is proposed through which the alanine substitution results in both the selectivity for critical metalloid nutrients such as boric acid while simultaneously restricting water flow through the ar/R selectivity filter. A mechanism involving two different rotameric states of the conserved arginine residue in this selectivity region is proposed to be responsible for the water-tight character of the pore. (Supported in part by NSF grant 1121465). 2206-Pos Board B343 Mechanism of Proton Transport of the M2 Proton Channel Studied by Constant pH Molecular Dynamics Wei Chen, Jana K. Shen. Department of Pharmaceutical Sciences, University of Maryland, Baltimore, MD, USA. The M2 protein of the influenza virus is a proton-selective homotetrameric channel. During virus entry, M2 is activated by the low pH of the endosome and transports protons into the virion, initiating viral uncoating. A histidine tetrad in the pore of the M2 transmembrane domain is responsible for pH activation and proton selectivity. A number of different mechanisms have been proposed for proton transport of M2 based on experimental and computational studies. To test these hypotheses, we applied the explicit-solvent continuous constant pH molecular dynamics method to study the pH-dependent conformational dynamics of M2 in explicit membrane. The calculated pKa values of the histidine tetrad are comparable to experimental values. The C-terminal openning of M2 became wider when pH was lowered. This work provides novel insight into the coupled protonation and conformational dynamics of the M2 proton channel. 2207-Pos Board B344 Proton Permeation in Ci-Hv1 Voltage-Gated Proton Channels occurs through a Proton Wire Involving Residues D160 and D222 and It is Modulated by N264 Amaury Pupo1, David Baez-Nieto1, Ester Otarola1, Osvaldo Yan˜ez2, Ariela Vergara-Jaque2, Wendy Gonzalez2, Karen Castillo1, Gustavo Contreras1, H. Peter Larsson3, Ramo´n Latorre1, Carlos Gonzalez1. 1 Centro Interdisciplinario de Neurociencia de Valparaiso, Valparaiso, Chile, 2 Universidad de Talca, Talca, Chile, 3University of Miami, Miami, FL, USA. Hv1 channels are integral membrane proteins with the capacity to selectively permeate protons in a voltage and pH-dependent manner. As Hv1 lacks a pore domain, permeation must occur through the voltage-sensing domain. Previous reports propose a permeation pathway consisting in a stable water wire which allows proton to permeate by means of a Grotthuss mechanism. Our molecular dynamics simulations do not support the formation of such stable water wire since it shows a dry zone around residue N264 in the wild type and in N264 mutants. Mutations of residues D222 and N264 affects single channel conductance (determined by non-stationary noise analysis) and selectivity, suggesting that both residues are involved in the permeation pathway. Quantum dynamics simulations performed in our model of the open Ci-Hv1 wt and in silico mutants suggest that permeation occur through a proton wire involving residues D160 and D222, a process modulated by N264. Supported by Beca de Doctorado Nacional para Extranjeros de Conicyt (A.P), FONDECYT Grants 1110430 (R.L.), 1120802 (C.G.); ANILLO Grant ACT1104 (C.G.); Postdoctoral Fellowships 3140590 (G.F.C.). CINV is a Millennium Institute. 2208-Pos Board B345 Free Energy Simulations of Ion Translocation through Voltage-Gated Proton Channel Hv1 Kethika Kulleperuma1,2, Susan M.E. Smith3, Thomas E. DeCoursey4, Regis Pomes1,2. 1 Department of Biochemistry, University of Toronto, Toronto, ON, Canada, 2 Molecular Structure and Function, Hospital for Sick Children, Toronto, ON, Canada, 3Department of Biology & Physics, Kennesaw State University, Toronto, GA, USA, 4Department of Molecular Biophysics & Physiology, Rush University, Chicago, IL, USA. The human voltage-gated proton channel (hHV1) is a transmembrane protein that is responsible for the selective permeation of protons across cell membranes in nasal mucosa, sperm, and white blood cells. hHV1 is a four-helix bundle (S1-S4) with anionic Asp112 on S1 forming a salt bridge with cationic Arg residues on helix S4 in the narrow region of the pore [Kulleperuma et al., J. Gen. Physiol. 141, 445-465 (2013)]. Mutation of Asp112 to Val abrogates channel properties. Unexpectedly, replacing Asp112 by a smaller neutral residue such as Ser turns HV1 into an anion selective channel that conducts Cl-, as does the double mutant D112V-V116S [Musset et al., Nature 480, 273-277 (2012); Morgan et al., J. Gen. Physiol. 142, 625-640 (2013)]. Although HV1 and its mutants exhibit drastic differences in ion permeation, the molecular 439a basis of proton selectivity in WT and anion selectivity in mutants remains unexplained. As the first step towards elucidating the charge selectivity of HV1, we perform molecular dynamics simulations with umbrella sampling to compute the free energy profile for the translocation of Naþ and Cl- ions through the pore of a homology model of HV1 and its mutants. The calculations are repeated in conformational states of the channel differing in the extent of hydration of the pore and in the relative arrangements of pore residues. Results show how ion solvation and electrostatic interactions with charged side chains in the pore lumen modulate the energetics of ion permeation in HV1 and suggest a structural basis for charge selectivity. 2209-Pos Board B346 Investigating the Potentiation Effect of 2-APB on CRAC Channels Xiaolan Xu, Sher Ali Syed, Tao Xu. Institute of Biophysics, CAS, Beijing, China. 2-aminoethyldiphenyl borate (2-APB) elicits both potentiation and inhibition effect on Ca2þ influx via CRAC channels. In this study we focused on understanding the underlying mechanism of its potentiation effect. We identified one key residue which are just located in the pore region, plays a vital role in the potentiating effect caused by 2-APB. Mutation of this residue with small side chain such as C, A, G, completely eliminate the potentiating effect, while mutation with large side chain such as M and I could generate 2-APB induced potentiation current as WT. Our results imply that the potentiation effect caused by 2-APB might has a close relationship with the change in the pore diameter. 2210-Pos Board B347 Slo2.x Potassium Channels are Involved in the Regulation of Heart Mitochondrial Function Charles Owen Smith. Biochemistry, University of Rochester, Rochester, NY, USA. Mitochondrial potassium channels (MKCs) are believed to be important in stress response in the heart. Volatile anesthetic preconditioning (APC) is a method of protecting the heart from ischemia-reperfusion injury which elicits evolutionarily-conserved protective signaling pathways that converge at the mitochondrial level. Work in C. elegans has focused attention on the Slo2 gene product as a transducer of APC effects on hypoxic survival and recent data from our lab demonstrate that this protective role is conserved in mammals. Slo2 in mammals has diverged into two paralogs, Slo2.1 (KCNT2; Slick) and Slo2.2 (KCNT1; Slack). These genes code for Naþ-activated Kþ channels and are highly expressed in brain, but their function in cardiomyocytes and/or mitochondria is unknown. Examination of these channels has been limited to pharmacologic profiling which is hampered by overlapping sensitivities and off-target effects of small molecules. Herein we employed novel genetic deletions of Slo2.1 and Slo2.2 double knockout, Slo2.x dKO, in mice to confirm the role of these potassium channels in APC and identify their role in endogenous cardiac mitochondrial function. Preliminary data obtained using Slo2.x dKO reveal novel metabolic and morphologic phenotypes, indicating a functional relationship between mitochondrial potassium channels and regulation of mitochondrial oxidative phosphorylation. These data demonstrate a role of the Slo2.x gene product in the regulation of cardiac mitochondrial function. 2211-Pos Board B348 Human Erythrocyte Mechano-Activated KD Channel A, a Kinetic Study of Intraburst Activity: Effect of Chlorpromazine Alejandro Mata1, Jesus G. Romero2. 1 Instituto de Biologia Experimental, Universidad Central Venezuela, Caracas, Venezuela, Bolivarian Republic of, 2Escuela de Biologia, Universidad Central Venezuela, Caracas, Venezuela, Bolivarian Republic of. Human red blood cells (hRBCs) have a mean life span of 120 days. However, little is known about this biological clock. We have presented evidence of the existence of the Human Erythrocyte Mechano-activated Kþ Channel A (HEMKCA), whose open probability (Po) depends on the pressure applied on the membrane. This channel shows a PKþ/PNaþz 100 with a mean conductance of 21.8 pS and it is modulated by Ca2þ(1)(2). We propose HEMKCA as the pressure sensor involved in the aging process of hRBCs at microcirculation level. Here we present a kinetic analysis for the HEMKCA burst activity in isolated membrane patches with 10 uM Ca2þ. In order to define the Tcrit we analyzed records with Po R 0.8 (90s), partitioned into windows of 10s. These records were classified as ‘‘low’’ (Po<0.9) and ‘‘high’’ (PoR0.9) activity. To determine the rate constants, interval durations were fitted by their corresponding probability density functions, assuming a Markov scheme with dead time of 430 us. We found that ‘‘High’’ activity requires at least two closed states (tau1¼0.2650.019; tau2¼12.2951.43), whereas ‘‘low’’ activity requires at least three closed states (tau1¼0.2550.017; tau2¼12.3551.45; tau3¼97.92525.92), with one state open in both cases 440a Tuesday, February 10, 2015 (‘‘high’’ tau¼28.4353.3; ‘‘low’’ tau¼22.251.08). Once we had the Tcrit (20.55 ms) we obtained the rate constants for intraburst activity with a C1O-C2 model: k 1-2 ¼ 182.44524.98; k 2-1 ¼4.5752.4; k 2-3 ¼ 196.33532.85; k 3-2 ¼ 4418.645299.37 (n ¼ 8). Finally we studied the effects of chlorpromazine, a known modulator of mechano-activated channels, on these rate constants and found a decrease in the k 3-2/k 2-3 relationship from 22.9753.39 to 10.6451.47 (p<0.05; n¼6). This model allows for formal kinetic studies of this novel channel. (1) (2005) Biophys J.88(1):593 (2) (2008) Biophys J.91(1):1101 2212-Pos Board B349 Mechanisms Underlying the Loss-Of-Functional Kir6.1 KATP Channel Mutations in Sudden Infant Death Syndrome Bi-Hua Tan1, Blaise Z. Peterson1, Ryan Li1, Tianyu Sun1, Sinisa Dovat1, Michael J. Ackerman2, Chunhua Song1. 1 Penn State College of Medicine, Hershey, PA, USA, 23Mayo Clinic College of Medicine, Rochester, MN, USA. INTRODUCTION: Hypoxia-induced apoptosis and arrhythmia are the important cause of sudden infant death syndrome (SIDS). ATP-sensitive Kþ (KATP) channels are known to provide a functional linkage between the electrical activity of cell membrane and metabolism. KCNJ8-encoded Kir6.1 KATP channel critically regulates vascular tone and cardiac adaptive response to systemic metabolic stressors, including sepsis. Previously, we identified two KCNJ8 mutations (E332del and V346I) in a large SIDS cohort that exhibited a marked loss-of-function phenotype and reduction of cell surface expression. Here we further investigate the mechanisms underlying the loss-of-functional Kir6.1 KATP channel mutations in SIDS. Methods and Results: A hemagglutinin (HA) epitope was inserted in an extracellular loop of Kir6.1 wild type (WT), Kir6.1-E332del and Kir6.1-V346I channels. HEK293 cells were co-transfected with cDNA encoding HAtagged Kir6.1-WT, HA-tagged Kir6.1-E332del or -V346I and SUR2A in a ratio of 1:1:2. Cell surface expression was assessed by Flow-cytometry with FITCconjugated anti-HA antibody. Apoptosis assays were performed on HEK293 cells transfected with IRES-GFP constructs containing Kir6.1-WT or mutant (E332del or V346I) with SUR2A. After staining with PE-Annexin V and 7-AAD, the apoptotic cells were measured by Flow-cytometry within gated GFP (þ) cells. Caspase-3/7 activity was measured with Apo-ONE Homogenous caspase 3/7 assay kit. The cell-counting studies showed that the cell surface expression of Kir6.1-WT was suppressed 40% to 70% when co-expressed with Kir6.1-E332del or Kir6.1-V346I. The apoptosis assay data indicated that the apoptotic ratio was increased significantly for E332del (38.9%) compared to WT (4.02%) and for V346I (11.2%) compared to WT (3.04%). The Caspase-3/7 activity was also increased 2.1 fold for E332del and 1.6 fold for V346I over WT. Conclusions: The loss-of-functional Kir6.1 KATP channel mutations found in SIDS display a dominant-negative effect on Kir6.1-WT channels and induce apoptosis in heterologous expression system. 2213-Pos Board B350 Studying Clustering of KcsA Channels using Single-Channel VoltageClamp Fluorescence Imaging Hugo McGuire, Rikard Blunck. Universite de Montreal, Montreal, QC, Canada. Protein oligomerization lies at the core of numerous biological processes, from cellular signal transduction to muscle contraction and cellular metabolism. It also notably drives the function of ion channels, as several subunits are often required to stabilize and gate specific ions through the conducting pore. In addition to these protein-protein interactions within ion channels, oligomerization or ‘‘clustering’’ of several ion channels has been observed. Although ion channel clustering is not generally thought to be a prerequisite for their physiological function, it has been suggested to modify the function of several ion channels. In this study, we optically observed clustering of single KcsA (E71A mutant) channels in planar lipid bilayer using single molecule fluorescence, while simultaneously measuring single channel currents. We found that clustering was not caused by direct protein-protein interactions but was mediated via microdomains induced in the lipid matrix. Interestingly, while KcsA clusters remained in the lipid bilayer, cooperative gating events with conductance levels multiple to the ‘‘normal’’ single channel events were often recorded. These coupled events were also observed in absence of a negatively charged phospholipid, which is believed to be required for KcsA activity. To understand the role of microdomains in coupled activity, we explored the physical properties of the lipid which could promote channel opening. Our findings show that lipids able to produce negative curvature in the lamellar liquid crystal phase (La) can induce channel activity, even without any negative charged lipid. We propose that the lateral pressure distribution of such lipid on the channel supports channel opening. This idea is in line with the cooperative gating of KcsA in the presence of clusters. The assembly of oligomers helps to overcome the energy barrier for opening by distributing the lateral pressure more favorably. 2214-Pos Board B351 Crystal Structure of Fluc, a Microbial Fluoride Channel Randy Stockbridge1, Ludmila Kolmakova-Partensky1, Akiko Koide2, Shohei Koide2, Simon Newstead3, Christopher Miller1. 1 Biochemistry, Brandeis University, Waltham, MA, USA, 2Biochemistry and Molecular Biology, University of Chicago, Chicago, IL, USA, 3 Biochemistry, University of Oxford, Oxford, United Kingdom. Widespread bacteria, archaea, unicellular eukaryotes, and plants possess fluoride channels, called Flucs, to export toxic environmental fluoride anion from the cytoplasm. These proteins conduct F- ion at ~10 pS, are >10,000fold selective for fluoride over chloride, and are constructed as extremely unusual antiparallel dimers. Here, we present the x-ray crystal structure of a Fluc channel from Bordetella pertussis, in complex with a pore-blocking FN3 domain ‘‘monobody.’’ The structure reveals a closed conformation of the protein with the predicted antiparallel architecture and an occluded hourglass-shaped pore at the subunit interface. 2215-Pos Board B352 Two-Sided Simultaneous Block of a F- Channel (FLUC) Daniel L. Turman1, Randy Stockbridge1, Christopher Miller2. 1 Brandeis University, Waltham, MA, USA, 2Brandeis University, HHMI, Waltham, MA, USA. The recently discovered family of F- channels, the Flucs, are highly selective for F- and function to rescue microorganisms from F- toxicity in acidic environments. Structurally, Fluc is a four pass transmembrane protein that assembles as a dual-topology homodimer. This architecture, where two Fluc subunits orient antiparallel with respect to each other, is unique among ion channels and requires a two-fold symmetry axis parallel to the membrane plane. The symmetry axis further suggests that Fluc may present identical interfaces on both sides of a membrane. In fact, using fibronectin III domain ‘‘monobody’’ blockers specifically selected from a phage display library for nano-molar binding affinity, it was shown that a single Fluc channel is blocked on both sides of a planar lipid bilayer [Stockbridge et. al. Nature Comm., in press]. This leaves open the question of whether the sites of block on either side of the channel can be occupied simultaneously. We approach this question by experimentally testing a two site block model O <¼> B1 <¼> B2 by analysis of monobody dependent block times in single channel electrophysiology recordings. In the case of symmetric block, this scheme postulates that the dwell times of the B1 and B2 composite blocked state should exhibit monobody concentration dependence. In addition, information about cooperativity, whether negative or positive, between the two blocking sites can be quantitatively determined. 2216-Pos Board B353 Regulation of ClC-3 Cl-/HD Transport and ‘‘Gating’’ Transients by Chloride Pathway Residues and External Protons Jeffrey Rohrbough, Fred S. Lamb, Hong-Ngan Nguyen. Pediatrics, Vanderbilt University School of Medicine, Nashville, TN, USA. We investigated ClC-3 ion transport properties by employing a ‘‘ClC-5/3’’ plasmid, consisting of the ClC-5 N-terminal (M1-A46) linked to the core ClC-3 protein. This ClC-3 protein is redirected to the plasma membrane of HEK cells. Functionally it exhibits rapidly activated, outwardly rectifying sustained ion currents (ISS) representing coupled Cl-/Hþ transport, and prominent ‘‘on/off’’ transient gating charge (Q) movements, as recently described for a ClC-3 mutant (Guzman et al, 2013, ACS Chem Neurosci). Interpreting Q to represent movements of unprotonated (ie, incompletely cycling) ‘‘Gluext’’/ E224, ClC-3 exhibits low transport efficiency compared to ClC-5 or ClC-4, which exhibit larger ISS and much smaller Q. Replacement of external Clwith SCN- increases ClC-3 currents by 3-4 fold, and reduces Hþ coupling by ~90%. Removal of a conserved tyrosine (Y630S, V) positioned at the intersection of the Cl-/Hþ pathways (Accardi et al, 2006, JMB) greatly increases currents and decreases Q. For Y630S, Hþ coupling is reduced by ~50%; SCN substitution decreases rather than increases current, and eliminates Hþ transport. An M568A mutant moderately impacts transport, increasing currents and decreasing Q by <50%. Reduction of external pH (pH 6 or 5) weakly inhibits ClC-3 ISS, but markedly reduces Q, and shifts the Q(V) relationship toward more positive voltages. These results are consistent with external protons inhibiting cycling by neutralization of Gluext. An endogenous Cl- current activated at pH5 (Iacid) has slow activation kinetics and lacks gating transients, is Tuesday, February 10, 2015 inhibited by 60-120 microM phloretin, and is decreased by ClC-3 expression. Thus, Iacid is readily distinguished from expressed ClC-3 transport. These data indicate that ClC-3 Hþ coupling and transport is influenced by anion interaction, and by external protons, as suggested for the bacterial ClCec-1. 2217-Pos Board B354 Energetics and Mechanism of Permeation across FNT Channels Kalina Atkovska, Jochen Hub. Institute for Microbiology and Genetics, Georg-August-University of Goettingen, Goettingen, Germany. The formate-nitrite transporters (FNTs) represent a widespread family of membrane proteins involved in the translocation of monovalent polyatomic anions, such as formate, nitrite and hydrosulfide. Recently solved structures of five of its members reveal a pentameric protein organization with remarkable structural similarity to aquaporins, which along with electrophysiological data suggest a channel-like permeation mechanism. Given the narrow and relatively hydrophobic nature of the permeation pore, the selectivity mechanism of FNTs remains poorly understood. The protonation state of the solutes, as well as the key residues involved in selectivity and gating have been debated, wherein a conserved histidine residue located central in the pore has been proposed to be crucial. We use atomistic molecular dynamics simulations to compute potentials of mean force for full permeation events of certain ionic and neutral solutes across four FNTs (nirC, HSC and two focA channels). Our analyses reveal a high permeation barrier (>75kJ/mol) for ions through the hydrophobic pore of all investigated channels. This barrier is significantly reduced or annihilated when either the central histidine residue, or the permeating solute are protonated. Moreover, we employ QM/MM calculations to investigate the proton exchange between said residue and the permeating solute, thus giving insight into the molecular details of permeation. In summary, the extensive calculations provide a detailed quantitative picture of the energetics of permeation of physiologically relevant solutes through all FNT subtypes with experimentally solved structure. 2218-Pos Board B355 Extracellular Chloride Regulates TMEM16A Gating Juan A. Contreras-Vite1, Silvia Cruz-Rangel1, Patricia Pe´rez-Cornejo2, H. Criss Hartzell3, Jorge Arreola1. 1 Institute of Physics, Univ. Autonoma de San Luis Potosi, San Luis Potosi, Mexico, 2School of Medicine, Univ. Autonoma de San Luis Potosi, San Luis Potosi, Mexico, 3Cell Biology, Emory University School of Medicine, Atlanta, GA, USA. The gating mechanism of TMEM16A, an essential subunit of the Ca2þ-activated Cl- channel, remains incompletely understood. Gating of TMEM16A channels is complex because elevation of intracellular calcium concentration ([Ca2þ]i), strong depolarizations (Vm), and high extracellular Cl or highly permeant anions seem to contribute. In the present work we show that in TMEM16A no gating currents can be detected, however, external Cl ions might play a role in voltage sensing because the kinetics and magnitude of current activation were dependent on [Cl-]e. The onset and offset of Cl- currents produced by 0.5 s depolarisations of cells bathed in 137 mM [Cl-]e followed a mono-exponential behaviour; a subsequent reduction of [Cl-]e to 10 mM diminished the conductance ~2-fold. These data suggest a single gating process regulated by extracellular Cl-. In contrast, when cells were exposed to 137 mM [Cl-]e the onset and offset currents elicited by 20s depolarisations exhibited fast and slow kinetics. Activation of the slow component resulted in large onset Cl- currents that did not reach steady-state at the end of the 20s pulse. This behaviour was abolished after activating TMEM16A with 5 mM [Ca2þ]i or after deleting four residues (448EAVK451) located in the first intracellular loop. Deleting four additional residues (444EEEEEAVK451) resulted in Cl- currents that decayed at the end of the 20 s depolarization. To explain our data we developed a 12-state gating model assuming that TMEM16A is activated by a direct, Vm-dependent binding of two Ca2þ ions. In this model external Cl- increases channel open probability by promoting stable Vm-dependent Ca2þ binding. Our model reproduced the gating behaviour of the TMEM16A currents in response to voltage, Ca2þ, and [Cl-]e. Thus, we conclude that external Cl stabilises Ca binding and promotes two gating modes in TMEM16A. Supported by grant 219949 from CONACyT. 2219-Pos Board B356 Activation of ATP Secretion via Volume-Regulated Anion Channels by Sphingosine-1-Phosphate in Raw Macrophages Philipp Burow, Manuela Klapperstu¨ck, Fritz Markwardt. JB Institute for Physiology, Martin-Luther University Halle, Halle (Saale), Germany. Whole cell ion currents were recorded in RAW 264.1 mouse macrophage cells by means of the voltage clamp technique in the whole cell configuration. 441a Outwardly rectifying anion currents were activated by sphingosine-1phosphate (S1P). The current reversal potential was shifted by replacement of extracellular Cl- by glutamate- but not when extracellular Naþ was substituted by Trisþ revealing that the S1P-induced current was mainly carried by anions. Similar currents were induced by hypotonic extracellular solution. The inhibition of the S1P-induced currents by hypertonic extracellular or hypotonic intracellular solution as well as the inhibitory effects of the anion channel blockers NPPB, tamoxifen and glibenclamide indicate that the anion current is mediated by volume-regulated anion channels (VRAC). The S1P effect was blocked by the blocker of the S1P receptor 1 subtype (S1PR1) W123 and by intracellular GDPbS which points to a signalling via S1PR1 and G-proteins. As ccytochalasin D diminished the action of S1P we conclude that the actin cytoskeleton is involved in the stimulation of VRAC. S1P as well as hypotonic extracellular solution induced a secretion of ATP from the macrophages which was blocked in both cases in a similar way by typical VRAC blockers. We suppose that the S1P-induced ATP secretion in macrophages via activation of VRAC constitutes a functional link between sphingolipid and purinergic signalling in essential processes such as inflammation, migration of leukocytes as well as phagocytosis and killing of intracellular bacteria. This work was supported by the Roux program of the Medical Faculty of the Martin Luther University Halle (FKZ 28/29). 2220-Pos Board B357 Positioning of the First Extracellular Loop of CFTR Has Significant Effects on CFTR Gating Daniel T. Infield, Guiying Cui, Christopher Kuang, Nael A. McCarty. Pediatrics, Emory University, Atlanta, GA, USA. The first extracellular loop (ECL1) of CFTR contains several residues involved in stabilizing the open state of CFTR. D110 is positioned on the side of ECL1 nearest to the CFTR pore, extracellular to several amino acids in the first transmembrane helix important for chloride permeation. In the present study, we utilized cysteine mutagenesis and electrophysiology to observe real time effects of chemical manipulation on D110C-CFTR and on a double mutant of D110C with K892C in ECL4, across the CFTR pore. Via whole Xenopus oocyte TEVC recording, we found that the reducing agent DTT increased the conductance of D110CCFTR 3.21 þ/ 1.16 fold and D110C/K892C-CFTR 13.0 þ/3.5 fold. Treatment of both variants after DTT with Copper (II) Phenanthroline quickly inhibited them to a similar extent (~75%). Single channel recordings without DTT showed that both mutants contain full conductance comparable to WTCFTR, but significantly decreased mean burst duration. We previously reported modification of D110C/K892C channels with DTT led to increased openings in multichannel patches, without apparent effects on single channel conductance or open burst duration, indicating that DTT likely breaks a closed-state linkage between D110C and K892C. Modification of D110C-CFTR with DTT resulted in an increase in mean burst duration from 154ms to 369ms. Finally, via TEVC, we found that 20uM Cadmium inhibited DTT-treated D110C-CFTR that reversed within 30s of washout, whereas D110C/K892C-CFTR was inhibited irreversibly in the same context. WT, K892C-, D112C/K892C-, and E115C/K892C-CFTR were unaffected by DTT or Cd2þ. We interpret our results to indicate the positioning of the pore-facing end of ECL1 is important for CFTR gating, and the more profound effects of DTT and Cd2þ on D110C/K892C-CFTR versus D110C-CFTR may also indicate ECL1 and ECL4 must separate during CFTR channel opening. Support: NIH 5R01DK56481 2221-Pos Board B358 Effects of the Connexin43 CT on Gap Junction Sub-Domain Organization and Myocyte-Fibroblast Interactions in the Injury Border Zone Emily L. Ongstad1, Robert G. Gourdie2. 1 Clemson University/ VTCRI, Roanoke, VA, USA, 2VTCRI, Roanoke, VA, USA. Phosphorylation of connexin43 (Cx43) has been shown to regulate gap junction (GJ) intercellular communication. A Cx43 carboxyl-terminus(CT) mimetic peptide (aCT1) increases GJ size via reduced Cx43/ZO-1 interaction in vitro and increases phosphorylation of Cx43 at aa S368 (Cx43pS368) in vitro and in vivo. We previously reported the effects of aCT1-treatment in a left ventricular (LV) cryoinjury model in vivo (Circ Res 2011;108(6):704-15). At 8-weeks after cryoinjury, histological assessment showed decreases in scar size and increases in collagen fibril uniformity in aCT1-treated hearts versus controls. Interdigitation of collagen and fibroblasts between myocytes in the injury border zone (IBZ) was reduced in treated hearts versus controls. Treated hearts exhibited a decreased propensity for induced arrhythmias (p<0.02) and an increase in Cx43pS368 in the IBZ which correlated with a tendency for Cx43 to be maintained at intercalated discs. To study the mechanistic effect of aCT1 on myocyte-fibroblast interactions via Cx43, we created a 3D heterocellular system to model the injury border zone 442a Tuesday, February 10, 2015 (IBZ) in vitro. Neonatal rat ventricular fibroblasts were seeded to form a shell around a core of aggregated neonatal rat ventricular myocytes (NRVMs) in 96well micromolds. After 72 hours, aCT1-treatment of aggregates induced an increase in heterocellular interactions via total Cx43 (p¼0.03) and Cx43pS368 (p¼0.01). Studies of an in vivo LV cryoinjury model were used to validate these data. Changes in myocyte-fibroblast interactions, GJ plaque size, and GJ sub-domain organization after aCT1 treatment were detected with confocal and super-resolution microscopy (both gSTED and STORM). Super-resolution microscopy enabled resolution of discrete phospho-S368 non-phosphorylated subdomains of Cx43 within GJ plaques. In sum, our results indicate the potential for Cx43 CT effects on heterocellular interactions via Cx43 in the IBZ and scar following myocardial infarction. 2222-Pos Board B359 Molecular Dynamics of Selective Sugar Permeation through the Connexin26 Channel Lyna Luo1, Angelo R. Rossi2, Andrew L. Harris2. 1 Department of Pharmaceutical Sciences, Western University of Health Sciences, Pomona, CA, USA, 2Pharmacology & Physiology, New Jersey Medical School - Rutgers University, Newark, NJ, USA. A signal property of connexin channels is the ability to mediate selective diffusive movement of molecules through plasma membrane, yet the movement of biological molecules through these channels has yet to be well-characterized in mechanistic or energetic terms. Different connexin channels have distinct molecular selectivities that cannot be explained on the basis of size or charge of the permeants; the forces that molecules experience within the pore determines which molecules are permeable and to what degree. The energetics of the movement of two derivatized sugars, one permeable and one impermeable, through an experimentally validated connexin26 (Cx26) structural model were explored using Hamiltonian Replica Exchange MD Umbrella Sampling (US/H-REMD) and Steered Molecular Dynamics (SMD), and associated analytic tools. Crucially, the Cx26 channel model, in explicit membrane/solvent, incorporates key posttranslational charge changes shown by Brownian Dynamics to be required to reproduce the electrical conductance characteristics of the native channel [Kwon J.Gen.Physiol. 138:4751]. The results show energy profiles consistent with experimental results. The energetic barriers extend through most of the pore length, rather than being highly localized, as in ion-specific channels. There is little evidence for binding within the pore. Force decomposition reveals how, for each test molecule, interactions with water and with the Cx26 protein vary over the length of the pore, and reveal a significant contribution of interaction with Kþ ions. The flexibility of pore width varies along its length, and the test molecules tend to widen the pore as they pass through. This work highlights factors involved in selective molecular permeation that may not be significant for atomic ions. The results suggest that this system can be used to explore the molecular basis by which connexin channels select among (potential) permeating molecules, and how mutations alter the permeation process. 2223-Pos Board B360 The Residues in the First Extracellular Domain Play an Important Role in Transjunctional-Voltage Dependent Gating and Unitary Channel Conductance of Cx50 Gap Junction Channels Xiaoling Tong1, Hiroshi Aoyama2, Swathy Sudhakar1, Honghong Chen1, Donglin Bai1. 1 Physiol. and Pharm., Western University, London, ON, Canada, 2Graduate School of Pharmaceutical Sciences, Osaka University, Osaka, Japan. Gap junctions (GJ) are intercellular channels that connecting the cytoplasm of neighboring cells. GJ channels formed by Cx50 and Cx36 show drastic disparity in their unitary channel conductance (gj) and transjunctional voltage-dependent gating (Vj-gating), but the important underlying molecular domains/residues are not clear. Experimental evidence showed that residues in the first extracellular domain (E1) of Cx50 likely line the GJ channel pore and may play an important role in determining gj and Vj-gating. We aligned the E1 sequence of Cx50 with that of Cx36 and found 10 different residues (4 out of 10 residues involve a change in charge). We generated a chimera Cx50Cx36E1, in which the E1 of Cx50 was replaced by the E1 of Cx36, and 4 point mutations in E1 of Cx50 (where a charge change occurs, i.e. G46D, D51M, E62N and E68R). Dual patch clamp study on the homotypic GJ channels formed by the chimera or the point mutants in N2A cell pairs indicate that the Cx50Cx36E1 channel showed little change in the Vj-gating properties, but displayed a significantly reduced gj. Surprisingly, homotypic G46D, E62N and E68R channels all increased gj and showed little change in Vj-gating properties, except E62N. D51M failed to form functional GJ channels. Our homology structure models of the chimera and the mutants indicate that the electrostatic properties of the pore-lining residues are a key parameter in facilitating ion permeation and play a role in the Vj-gating properties of Cx50 and possibly other GJ channels. 2224-Pos Board B361 Residues Involved in Cx26 Hemichannels Voltage Dependent Gating Bernardo I. Pinto, David Baez-Nieto, Amaury Pupo, Agustin Martinez, Ramon Latorre, Carlos Gonzalez. Centro Interdisciplinario de Neurociencias, Valparaiso, Chile. The most well-known role of Connexins (Cxs) is to communicate the cytoplasm of two adjacent cells forming a gap junction channel in the apposition zone. On the other hand, Cxs form the so called ‘‘hemichannels’’, a fully functional voltage-gated non-selective ion channel, which present two gating mechanism called ‘‘slow’’ and ‘‘fast’’ gating. Hemichannels consist of 6 monomers, each one composed of four transmembrane domains. Despite the fact that hemichannels are voltage-activated, they lack the classical voltage-sensing domain described for canonical voltage-gated channels. Furthermore, the molecular determinants associated to the voltage activation in connexins hemichannels are still elusive. In this work we search for the molecular determinants associated to voltage detection in human Cx26 hemichannels. Using two electrode voltage-clamp in Xenopus laevis oocytes, we study how the steady-state conductance-voltage (G/V) relationship and kinetics of gating are affected by the neutralization of different charged residues located within the first transmembrane domain. We have found that Cx26 conductance-voltage curve shows an apparent number of gating charges (zd) of 1.4 e0 for the slow gating mechanism. On the other hand, we obtained a zd of 3.9 e0 for the fast gate, which is in agreement with the zd of Cx26 gap junctions. A simple three-state closed-open1-open2 kinetic model is able to account for the channel voltage dependence. Furthermore, our data shows that neutralizing the K41 residue (K41N) increases the zd; of the slow gate to 2.5 e0, and decreases the zd of the fast gate to 2.3 e0. This work is supported by FONDECYT Grants 1110430 (R.L.), 1120802 (C.G.), 1130855 (A.M.); ANILLO Grant ACT1104 (C.G.); Beca para Estudios de Magı´ster en Chile An˜o 2014 (B.P.); CINV is a Millennium Institute supported by the Millennium Scientific Initiative of the Ministerio de Economı´a, Fomento y Turismo. 2225-Pos Board B362 Understanding the Conformational Dynamics of the Porin Occk5 Karunakar Reddy Pothula. The outer membrane of Gram-negative bacteria contains various porins to permit the passage of water soluble molecules into the periplasm of bacteria. Compared to the Gram-negative bacterium Escherichia coli, the pathogenic organism Pseudomonas aerugionsa mainly contains substrate-specific, relatively small-sized porins making the transport of various substrates through its outer membrane difficult. Small molecules containing carboxylic acid group are taken up by the members of the Occ family (1). Thus, understanding the structure-function relationship of these porins is crucial to rationally design new antibiotics with better permeability. Recent electrophysiological experiments have shown that OccK5 - a member of the Occ family of proteins - exists in two conducting substates, i.e., O2 and O1 (2). Our goal is to study the conformational dynamics of OccK5 porin using molecular dynamics (MD) simulations (3). To this end, accelerated MD simulations have been used to characterize the structural and dynamic properties of the OccK5 porin. Using accelerated simulations, transitions between O2 and O1 substates have been achieved. Our study provides the first atomistic view of the transition between O2 and O1 substates in OccK5. Particularly, the role of specific arginine side chain conformations in defining the different substates of OccK5 is described. To further confirm these findings a temperature-dependent single channel analysis has been performed. Experimental data reveal kinetic and thermodynamic constants of the transitions between the observed major sub-states and probable transition intermediates. References (1) Tamber, S., et al.; J. Bacteriol., 189:929 (2007). (2) Liu, J., et al.; Biochemistry, 51:2319 (2012). (3) Modi, N., et al.; Nanoscale, 4:6166 (2012). 2226-Pos Board B363 Cyclodextrin Interaction with Specific Channel CymA from K. Oxytoca Satya Prathyusha Bhamidimarri1, Jing Lu1, Jigneshkumar Dahyabhai Prajapati1, Ivan Barcena Uribarri1, Bert van den Berg2, Ulrich Kleinekathoefer1, Mathias Winterhalter1. 1 Molecular Biophysics, Jacobs University Bremen, bremen, Germany, 2 Institute for cell and molecular biosciences, University of New Castle, New Castle upon Tyne, United Kingdom. The outer membrane acts as a selective uptake barrier in Gram negative bacteria. It contains protein channels (porins) which provide an entry pathway for Tuesday, February 10, 2015 hydrophilic molecules like small nutrient molecules and b-lactam antibiotics. However the CymA channel is known to take up cyclodextrin molecules giving bacteria the ability to survive on cyclodextrins. Hence understanding uptake of these molecules via porins is vital to comprehend the transport mechanism across the cell membrane. Electrophysiology forms a promising approach to study the permeation of molecules across outer membrane and thereby understanding molecular interactions with the channel. Here we present cyclodextrin interaction studies of CymA from K. oxytoca using single channel electrophysiology. Detailed single channel analysis revealed inherent asymmetric gating characteristics of the channel. Analysis of the ion current reduction through CymA in presence of cyclodextrin led revealed kinetic parameters of substrate binding. To further elucidate the affinity sites of substrate to the channel, mutation of certain channel residues has been performed. An altered channel gating behaviour is observed. To obtain an atomistic view we complement our studies with all-atom molecular dynamics simulation to study the various conductance states of the channel in the absence of cyclodextrin and to get molecular insight into the uptake of cyclodextrins as well. References: 1. Orlik F, Andersen C, Danelon C, Winterhalter M, Pajatsch M, et al. 2003. Biophysical journal 85:876-85 2. Pajatsch M, Andersen C, Mathes A, Bock A, Benz R, Engelhardt H. 1999. The Journal of biological chemistry 274:25159-66 2227-Pos Board B364 Mimicking Biology with Nanomaterials: Carbon Nanotube Porins in Lipid Membranes Aleksandr Noy. Lawrence Livermore National Laboratory, Livermore, CA, USA. Living systems control transport of ions or small molecules across biological membranes using ion channels that form highly efficient and selective pores in lipid bilayers. Although bottom-up synthesis and top-down fabrication could produce pores of comparable size, an unresolved challenge remains to build nanopore scaffolds that fully replicate transport properties of membrane channels. We will show that pores formed by ultra-short carbon nanotubes (CNTs) assembled in the lipid membranes have transport properties that come remarkably close to that goal. These CNT porins can transport water, protons, small ions, and DNA and their ion-rejection properties can be controlled by the charge at the pore mouth. Interestingly, these pores also display the stochastic ‘‘gating’’ behavior common for biological ion channels. Overall, CNT porins represent a simplified biomimetic system that is ideal for studying fundamentals of transport in biological channels, and for building engineered mesoscale structures, such as artificial cells. 2228-Pos Board B365 Understanding the Translocation of Fluoroquinolones through OmpC using the Metadynamics Jigneshkumar D. Prajapati1, Harsha Bajaj1, Matteo Ceccarelli2, Mathias Winterhalter1, Ulrich Kleinekatho¨fer1. 1 School of Engineering and Science, Jacobs University Bremen, Bremen, Germany, 2Dipartimento di Fisica and Istituto Officina dei Materiali/CNR, U






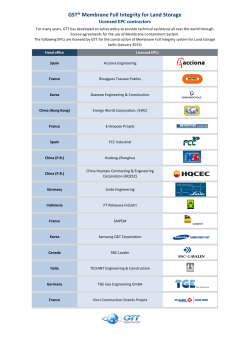



© Copyright 2025